基于ITS 基因序列的我国东南沿海等边浅蛤遗传多样性及遗传结构研究
2023-06-28李重黄继顾忠旗叶莹莹
李重,黄继,顾忠旗,叶莹莹
(1.浙江海洋大学国家海洋设施养殖工程技术研究中心,浙江舟山 316022;2.嵊泗县海洋科技研究所,浙江嵊泗 202450)
等边浅蛤Macridiscus multifarius俗称沙蛤[1]。该物种生活于我国沿海潮间带的中潮、低潮带至浅海的泥沙海底,另外,其在日本、韩国、朝鲜、越南、印度和印度尼西亚也有分布[2]。等边浅蛤贝壳呈三角卵圆形,壳质坚厚、重,其味道鲜美及营养丰富,由于具有一定的经济和食用价值而成为我国广泛养殖的海水贝类之一[3]。目前关于等边浅蛤的分子生物学相关研究较少,其中夏立萍等[3]基于高通量测序技术对等边浅蛤的线粒体全基因组进行了测序并进行了系统进化研究。徐鸣[4]利用线粒体16S rRNA 基因、Cytb基因、COX3基因作为分子标记分析了我国沿海等边浅蛤的遗传多样性及遗传结构。于瑞海等[5]取活的等边浅蛤鳃组织为材料,对等边浅蛤线粒体核型进行了分析研究。针对等边浅蛤核糖体ITS基因标记的研究还未有报道。
核糖体DNA(nuclear ribosomal DNA,rDNA)由于种内基因的流动时常表现出非常高的同源性,在种群间仍保持着多种程度的变异[6]。在rDNA 基因中,5.8S rDNA 和28S rDNA 基因间隔序列(internal transcribed spacer)即ITS基因序列非常保守,且在种群间表现比较高的差异。近年来常被用于亲缘关系鉴定[7]、系统进化[8]及种群遗传结构[9]等方面的研究。此外,这一技术也广泛应用于动植物病原菌检测和诊断[10]。
近些年来,由于海洋污染与过度捕捞,等边浅蛤野生资源的保护正面临巨大挑战。本研究以核糖体DNA的ITS基因序列为分子标记,对我国东南沿海4 个不同地理群体的等边浅蛤进行了遗传多样性及遗传结构分析,以补充等边浅蛤群体遗传相关领域研究的不足,为我国等边浅蛤种质资源的保护研究提供理论依据。
1 材料与方法
1.1 样品采集
本研究所用等边浅蛤采集于4 个沿海地区,分别为浙江舟山(ZS)、浙江南麂岛(NJ)、福建漳浦(ZP)、广西北海(BH),如图1 所示。在每个地点选取多处采集点并随机取样,采集好的样本放入冰盒保存,在实验室内解剖,取其闭壳肌组织保存于无水乙醇中,并采用改良盐析法[11]提取总DNA,用琼脂糖凝胶电泳进行质量检测,后置于-20 ℃下保存备用。
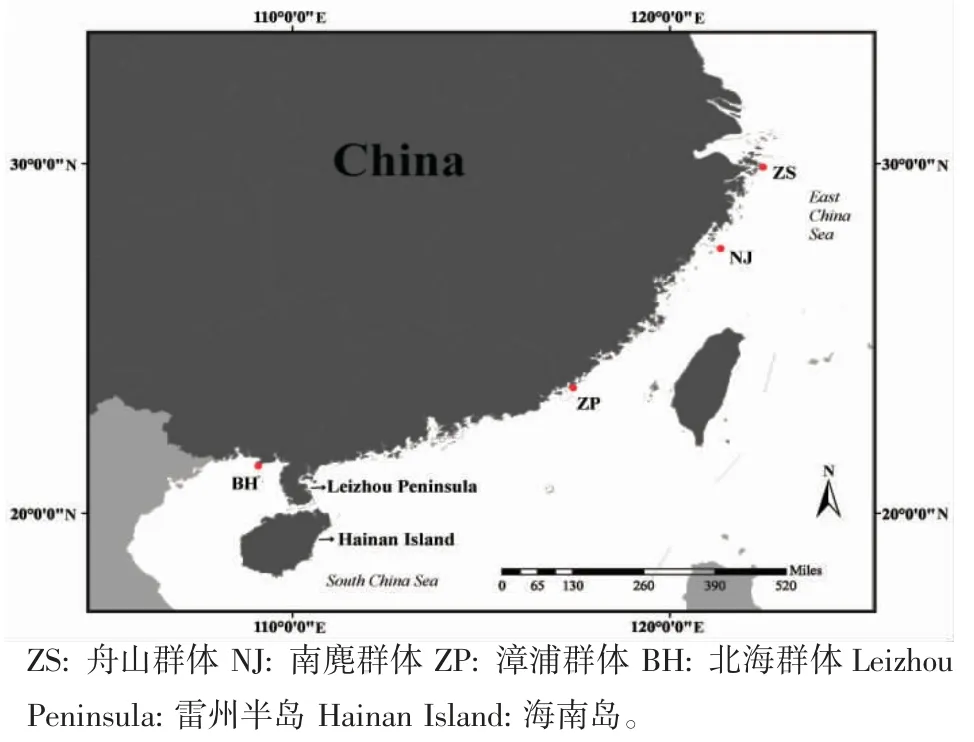
图1 等边浅蛤群体样本采集点Fig.1 Collection locations of M.multifarius populations
1.2 核糖体ITS 序列的扩增
等边浅蛤核糖体ITS 序列的扩增引物,来源于通用引物[12],序列如下:ITS 5.8S:5’-GCATCGATGAAGAACGCAGC-3’,ITS 18S:5’-CGCCGTTACTAGGGGAATCCTTGTAAG-3’。PCR 体系:2×TaqMix 酶12.5 μL、模板DNA 0.5 μL、上下游引物各1 μL(引物浓度为10 mmol·L-1)与10 μL ddH2O(up to 25 μL)。PCR 反应条件:在96 ℃下完成2 min 的预变性,30 s 变性,53 ℃下复性30 s,73 ℃延伸90 s,在完成35 个循环反应后,在73 ℃延伸10 min。PCR 产物经1.5%琼脂糖凝胶电泳检测后,将质量符合后续实验要求的PCR 产物送至杭州擎科生物技术有限公司进行双向测序。
1.3 序列分析及系统发育树构建
通过MEGA v7.0[13]软件对测序获得的ITS序列进行比对矫正,并对齐全部序列片段,为了进一步确定样本的准确性,以及所得片段的准确位置,在NCBI 数据库中的BLAST 工具上(https://blast.ncbi.nlm.nih.gov/Blast.cgi)对比序列结果[14]。利用MEGA v7.0[13]基于Kimura 2-parameter 模型,采用邻接法(neighbor-joining method)构建系统发育树,设置参数自举检验值(bootstrap)1 000 次重复检验各分支。利用软件DNASP[15]计算各类遗传学参数,包括单倍型数、单倍型多样性(h)和核苷酸多样性(π)。利用Arlequin3.5[16]软件计算遗传分化系数(Fst),并计算Fst的P值,同时使用Arlequinv3.5[16]进行分子方差分析(AMOVA)、并在Tajima′sD与Fu′sFs模型下,检测数据是否符合中性进化理论。利用软件Network v5.1[17]基于中介邻接法(median-joining method)构建ITS序列标记的单倍型网络图。
2 结果与分析
2.1 遗传多样性分析
对得到的81 个等边浅蛤样本进行PCR 扩增测序后,我们获得了长度为420 bp的ITS基因序列(NCBI登录号分别为:ON870718-ON870798)。数据分析显示在获得的ITS基因片段样本中含有31 个单倍型(表1)。其中有8 个共享单倍型,Hap-1 被舟山、南麂岛、漳浦群体共享,Hap-2 被舟山、北海、漳浦群体共享。Hap-13 被舟山和南麂岛群体同时共享,Hap-12、Hap-16、Hap-19、Hap-18、Hap-20 被漳浦和南麂岛群体同时共享。其余单倍型均为各个群体所特有的单倍型。
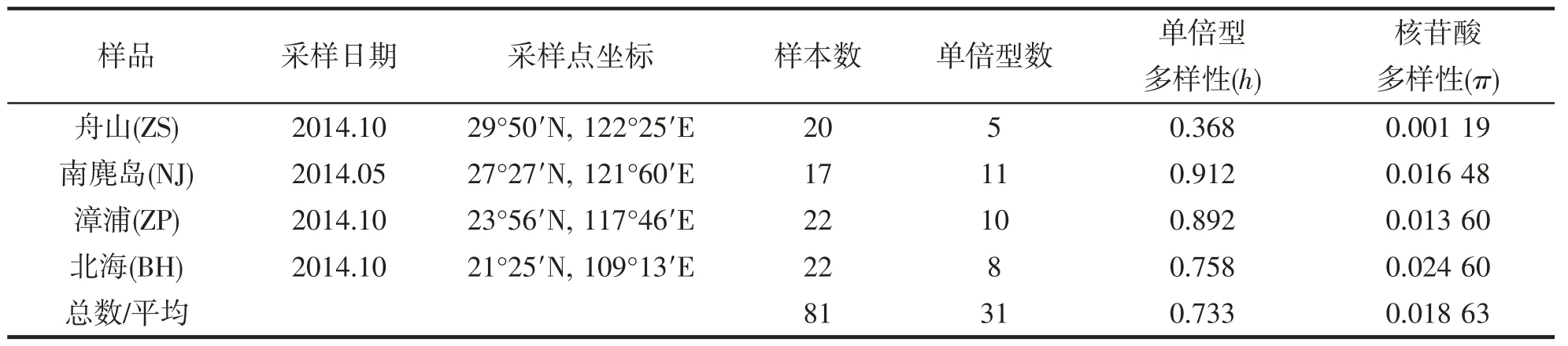
表1 基于ITS 标记的等边浅蛤4 个群体的遗传学参数Tab.1 Genetic diversity parameters of partical ITS gene in 4 populations of M.multifarius
等边浅蛤4 个群体的ITS序列的遗传学参数如表1 所示。其中平均单倍型多样性处于中等偏高水平(h=0.733),南麂岛和漳浦群体单倍型多样性相对较高(h=0.912和h=0.892),舟山群体单倍型多样性最低(h=0.368)。在核苷酸多样性水平的数据上,4 个群体的平均值偏低(π=0.018 63),其中舟山群体的核苷酸多样性水平最低(π=0.001 19)。舟山群体的单倍型多样性和核苷酸多样性偏低,可能与样本采集点过于集中有关。
2.2 群体遗传结构的研究
如表2 所示,在ITS基因序列中遗传分化系数Fst的范围在0.033~0.254 之间,其中北海(BH)和舟山(ZS)之间Fst最大值为0.254(P<0.05),北海(BH)和南麂(NJ)之间Fst为0.234(P<0.05),北海(BH)和漳浦(ZP)之间Fst为0.214(P<0.05)。表内数据显示,北海(BH)群体与其他群体之间存在中等遗传分化,其他群体间遗传分化较小且不显著(P>0.05)。
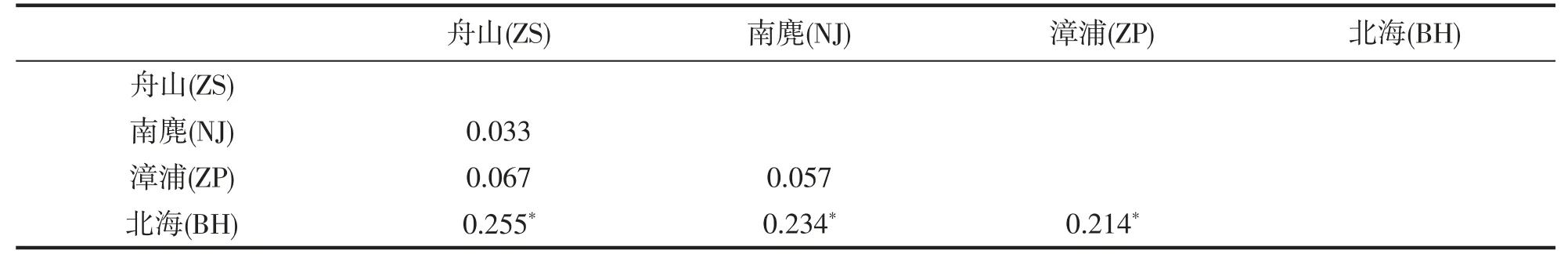
表2 基于ITS 序列的等边浅蛤群体遗传分化Tab.2 Estimates of population genetic differentiation based on ITS gene for the M.multifarius
基于4 个等边浅蛤群体的ITS基因构建的系统进化树如图2 所示,该系统进化树总体分为2 大支,北海(BH)群体为单独的1 支,其他群体在另1 大支上。
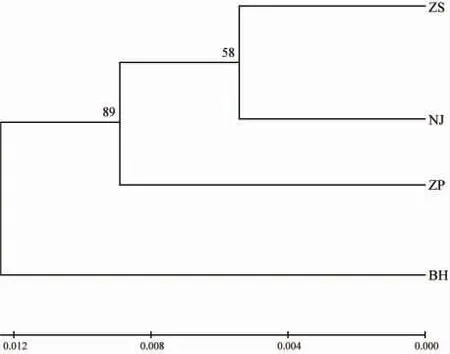
图2 等边浅蛤4 个群体ITS 序列的NJ 系统发育树Fig.2 The NJ phylogenetic tree of four populations of M.multifarius based on ITS sequences
图3 为等边浅蛤群体的单倍型网络图,其中Hap-1 被舟山(ZS)、南麂(NJ)、漳浦(ZP)3 个群体共享,位于中央。北海(BH)群体的单倍型相比于其他群体的单倍型出现较为明显的地理分支,这表明北海(BH)群体相较于其他群体发生了遗传分化,这与系统发育树和Fst值结果一致。

图3 等边浅蛤4 个群体的ITS 序列单倍型网络图Fig.3 The haplotype network of four populations of M.multifarius based on ITS sequences
2.3 群体历史动态分析
表3 为AMOVA 结果,4 个等边浅蛤群体间和群体内遗传变异分别占31.13%和69.87%,表明群体内遗传分化比例远大于群体间遗传分化比例。中性检验结果(表4)显示,4 个等边浅蛤群体的ITS序列的Tajima′sD值均不显著(P>0.05),Fu′sFs值总体未达到显著水平(P>0.05)。这表明这4 个等边浅蛤群体符合中性进化,无群体扩张现象发生。

表3 等边浅蛤4 个群体ITS 序列遗传差异的分子方差分析(AMOVA)Tab.3 AMOVA analysis among 4 populations of M.multifarius based on ITS sequences
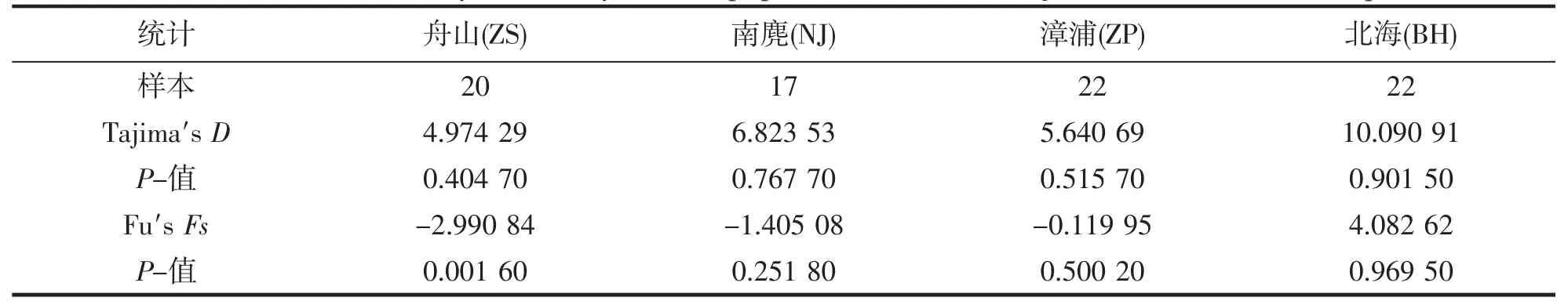
表4 等边浅蛤4 个群体基于ITS 序列的中性检测结果Tab.4 Selective neutrality test analysis of 4 populations of M.multifarius based on ITS sequences
3 讨论
3.1 遗传多样性
遗传多样性是衡量物种适应复杂环境的潜力的重要指标[18]。相对来讲,遗传多样性水平越高的种群,其对环境的适应能力就越强。所以研究物种遗传多样性有助于我们了解物种的进化潜力,对于物种多样性的保护具有重要的意义[19]。本研究基于核糖体DNA的ITS基因序列探究了我国东南沿海4 个不同地理位置等边浅蛤群体的遗传多样性及遗传结构。衡量物种遗传多样性是否丰富的重要的指标包括核苷酸多样性和单倍型多样性[20],核苷酸多样性和单倍型多样性越高,遗传多样性越大[21]。在本研究中,4 个群体的等边浅蛤一共检测到31 个单倍型,其平均单倍型多样性为0.733,核苷酸多样性为0.018 63,4 个等边浅蛤群体总体呈现较高的遗传多样性。等边浅蛤通过体外受精的方式繁育个体、浮游幼虫的生活范围较大,且生殖能力比较强,这些都是等边浅蛤遗传多样性比较高的原因[22]。这些现象在海洋贝类中也普遍存在[23],倪刚[24]基于线粒体和核DNA 分子标记研究了4 种广布性贝类系统地理学,同时发现青蛤Cyclina sinensis、泥蚶Tegillarca granosa、中国蛤蜊Mactra chinensis、四角蛤蜊Mactra veneriformis这4 种贝类遗传多样性都较为丰富。尤仲杰等[25]采用随机扩增多态性(RAPD)技术,对5 个西施舌Mactra antiquata自然群体的遗传多样性进行了研究,发现我国5 个西施舌群体的多态位点比例平均值为78.97%,平均遗传杂合度为0.309,处于较高的遗传多样性水平。
中性检验数据显示4 个等边浅蛤群体没有发生过群体扩张,所以等边浅蛤遗传多样性的高水平很可能与其体外受精以及生活环境周围的洋流与潮流影响相关[26]。此结果与田云方等[27]利用线粒体COI基因研究丽文蛤Meretrix lusoria群体遗传多样性的结论相似。
3.2 群体遗传结构
群体遗传结构是生物多样性的保护单元,在进化上具有重要意义。Fst值是衡量一个物种遗传分化指数高低的指标,Fst值越大,说明群体之间的遗传分化水平就越大[28]。一个物种近亲繁殖程度的高低也可以通过Fst值来反映[29]。本研究中Fst值的研究数据显示,北海群体与舟山、南麂岛、漳浦群体之间存在显著遗传分化,而舟山、南麂岛、漳浦群体间遗传分化较小且不显著。NJ 系统进化树显示,北海群体为单独的1支,其他群体聚为另1 大支。单倍型网络图同样可以反映群体的遗传结构关系,从单倍型网络图来看,北海群体的单倍型相比于其他群体的单倍型出现较为明显的地理分支,这与Fst值以及NJ 进化树显示的结果一致。
海峡隔离作用和洋流活动会影响海洋生物的遗传结构[30],王景博等[31]采用AFLP 分子标记对中国沿海的6 个野生缢蛏Sinonovacula constricta群体的遗传多样性和遗传结构进行研究,发现由于海南岛与雷州半岛的地理隔离作用,北海的野生缢蛏群体与其他群体间存在显著的遗传分化[27]。本研究中等边浅蛤样本采样地的4 个地理位置中北海位于北部湾海域,北海群体与其他群体间被琼州海峡隔离。此外,等边浅蛤在其幼虫阶段会以浮游方式生活[28],其浮游期较短,为10 d 左右[32],由于雷州半岛的隔离作用,北海地区等边浅蛤群体生活在较为单一的环流之中,所以北海群体与其他3 个群体间基因交流不大,与其他3 个群体间分化明显。
4 小结
本文对我国东南沿海4 个地理群体的等边浅蛤进行群体遗传学研究,结果显示北海群体和舟山、南麂岛、漳浦群体间存在显著遗传分化,这与等边浅蛤浮游幼虫期较短、北海地区等边浅蛤群体生活环境周围的洋流影响以及海南岛与雷州半岛的地理隔离作用有关。目前随着海洋经济的发展,海洋污染及海洋生物多样性衰减等问题愈发突出,等边浅蛤作为我国沿海潮间带的一种优质贝类,不仅是一个经济物种,其作为食物链中的一环,在生态系统的物质循环和能量流动也扮演着重要的角色。对等边浅蛤群体遗传多样性及遗传结构的研究,将有助于更好的指导等边浅蛤群体资源保护与开发利用,同时也对保护浅蛤属的物种多样性具有重要的意义。
