白塞病合并中枢性尿崩症1例并文献复习
2023-06-28黄林丽朱丰林刘春红杰东甘建平吴斌
黄林丽 朱丰林 刘春红 杰东 甘建平 吴斌
[摘要] 白塞病是一种反复发作的炎症性自身免疫性疾病,该病可累及黏膜、皮肤、眼睛、消化道、神经、心脏、肺脏、肾脏、关节、附睾等多系统损害。中枢性尿崩症是一种临床综合征,临床主要表现为多饮、多尿、烦渴、低渗透压尿。白塞病合并中枢性尿崩症在临床发病率极低,临床易误诊、误治。笔者结合相关文献对白塞病合并中枢性尿崩症的临床表现、机制、诊断、治疗进行总结,以提高临床对该病的认识。
[关键词] 白塞病;中枢性尿崩症;临床表现;诊疗
[中图分类号] R593.2 [文献标识码] A [DOI] 10.3969/j.issn.1673-9701.2023.12.033
白塞病(Behcet's disease,BD)是一种反复发作的炎症性自身免疫性疾病,其发病机制目前仍不明确,可能与遗传、免疫异常、感染、生活环境等因素密切相关。白塞病好发于30~40 岁的青年人,40岁以后发病率较罕见[1],该病可导致黏膜、皮肤、眼睛、消化道、神经、心脏等多系统损害[2]。中枢性尿崩症(central diabetes insipidus,CDI)是一种临床综合征,其是由于下丘脑或垂体后叶血管加压素能神经元的丧失或功能受损,导致精氨酸加压素的合成和(或)分泌受损,精氨酸加压素缺乏又导致无法浓缩尿液和过多的肾脏水分流失,最终导致以低渗性多尿伴代偿性口渴为主的临床表现[3]。多种病因可引起CDI 的神经元破坏,通常是由肿瘤对血管加压素能神经元的解剖学破坏、创伤性脑损伤或神经外科干预导致的创伤性损伤及精氨酸加压素分泌神经元的自身免疫性破坏三种病因发展而来。此外,在儿童时期还会发生由精氨酸加压素基因突变引起的家族性中枢性尿崩症[4]。目前为止关于BD 合并CDI的报道非常少见,在临床中极易误诊、误治,本文报道1 例该病患者,以提高临床医生对此类疾病的认识。
1 病例资料
患者,女性,31 岁,2021 年7 月29 日就诊,以反复口腔、外阴溃疡1 年余为主诉。患者1 年余来无明显诱因出现反复口腔溃疡及外阴溃疡3 次以上,并呈进行性加重。在外院查免疫球蛋白E134.00IU/ml,红细胞沉降率27mm/h,抗内皮细胞抗体1:320P 阳性。结核T 细胞检测、艾滋病毒筛查、血管炎抗体谱、抗核抗体谱、抗β2 糖蛋白1 抗体测定阴性,阴道镜检查提示外阴白色病变,全身PET/CT检查未见异常。患者为进一步治疗,来重庆市中医院风湿病科就诊,门诊以“白塞氏病?”收住入院。入院症见:患者口腔、外阴溃疡伴疼痛明显,口干、眼干、小便量多,每日约3~4L,无尿急尿痛,无眼红眼痛、视力减退,无结节性红斑、丘疹性脓疱。无外伤史,无家族遗传史。入院查体:生命体征平稳,心肺腹未见明显异常。口腔及牙龈黏膜可见数个黄豆大小溃疡,界清,溃疡面为白色,溃疡周围黏膜充血呈红晕状。外阴可见散在粟粒样大小不等溃疡,局部溃疡面可见白色分泌物。入院查体:红细胞沉降率30mm/h,血常规、尿液分析、粪便常规、肝肾功能、血糖、血脂、电解质、D-二聚体、大便隐血试验、心肌酶谱、抗CCP 抗体、类风湿因子、C 反应蛋白、补体C3、C4、免疫球蛋白、人类免疫缺陷病毒初筛、结核抗体、血清IgG4 未见异常。唇腺活检及免疫组化示:少量淋巴细胞浸润(<50 个/4mm2)、IgG4 阴性。针刺反应阴性。
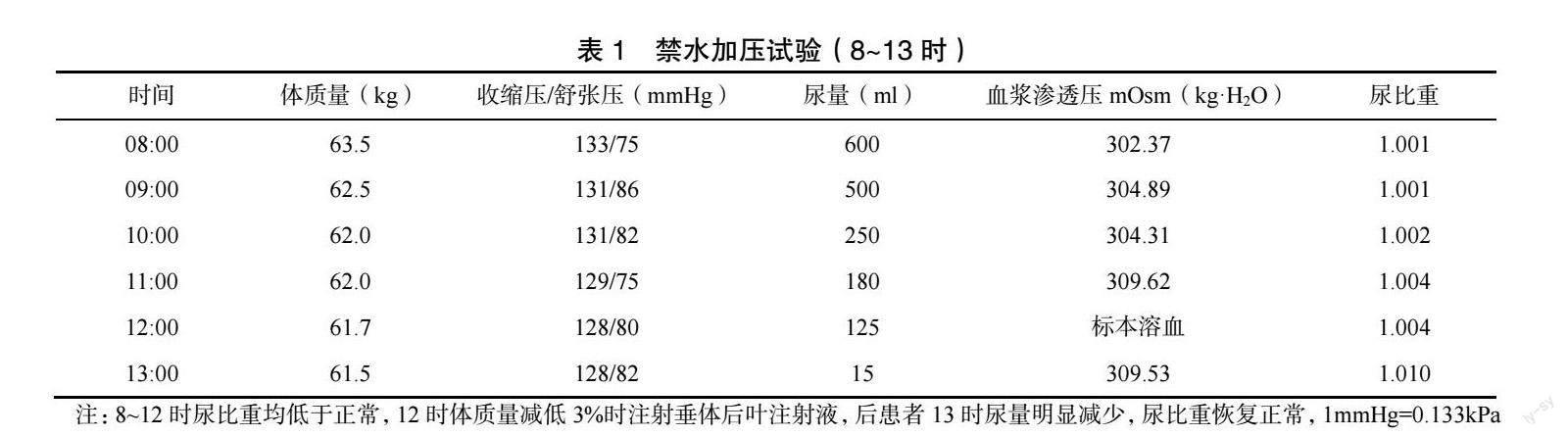
患者病程1 年余,反复出现口腔、外阴溃疡3次以上,并呈进行性加重。根据BD2014 年欧洲国際诊断标准,结合患者症状、查体及相关检查,并排除干燥综合征、副肿瘤综合征、结核感染、免疫球蛋白G4 相关性疾病,故诊断为BD。治疗上,予甲泼尼龙片(批准文号:国药准字H20020224,生产单位:天津天药药业股份有限公司,规格:4mg)每次12mg,口服,1 次/d;沙利度胺片(批准文号:国药准字H32026129,生产单位:常州制药厂有限公司,规格:25mg)每次50mg,口服,1 次/d;甲氨蝶呤(批准文号:国药准字H22022674,生产单位:通化茂祥制药有限公司,规格:2.5g)片7.5mg,口服,1 次/周;辅以康复新液外用,予药物治疗后,患者尿多情况仍存在且日益加重,还出现口渴烦躁,多饮,每日饮水量4~6L 左右,尿量与饮水量相当。复查尿液分析提示尿比重为1.002,转入内分泌科,查生长激素、促肾上腺皮质激素、性腺激素、催乳素、甲状腺激素正常,尿液分析提示尿比重为1.001;鞍区磁共振增强成像:①垂体上垂体柄旁T1WI 高信号,考虑神经垂体异位②垂体右份小结节,微腺瘤待排。禁水加压试验阳性。结合患者BD 病史、症状及相关检查、试验,并排除肾性尿崩症、原发性烦渴、药物不良反应等其他导致尿崩症可能性疾病,诊断考虑BD 合并CDI。治疗方面,在前方案基础上加予醋酸去氨加压素片(批准文号:国药准字H20090426,生产单位:辉凌制药有限公司,规格:0.1mg)每次0.1mg,3 次/d,治疗9d 后,患者尿崩症好转。后患者至本院门诊规律复诊,持续逐渐减量甲泼尼龙片剂量,其余药物剂量不变,现患者无明显口腔、外阴溃疡及眼干,烦渴、多饮、多尿控制可。门诊多次复查血沉、尿比重正常。
2 讨论
2.1 BD 合并CDI 尿崩症的临床表现
BD 临床上常见症状是复发性口腔及生殖器溃疡、眼部炎症及皮疹。BD 的口腔溃疡通常表现很轻微,它们的直径通常<1cm,位于脸颊、嘴唇、舌头和牙龈的黏膜内层[1]。生殖器溃疡通常位于男性阴囊和女性大阴唇上,约2/3 的患者出现较大的瘢痕表现。眼部病变在BD 中的发病率约为45%~90%[5],包括结膜溃疡、巩膜外层炎、葡萄膜炎、神经眼科表现、视网膜血管炎。其中葡萄膜炎和神经眼科表现是威胁视力的疾病,预后较差。BD患者经常出现丘疹脓疱性病变,研究发现其病变常见于腿部,会持续到老年[1]。除了上述常见症状外,BD 还易累及合并关节、肌肉、血管、神经系统、胃肠道、附睾等其他疾病表现。BD 累及的关节炎通常是一种复发性、非变形性单关节炎或少关节炎[6]。当累及肌肉时,则主要表现为纤维肌痛,给患者带来极大的痛苦。BD 也会累及动静脉形成血管炎,主要包括视网膜血管炎、肠系膜动脉血管炎、血管周围炎症和大脑中的毛细血管或静脉受损,及中性粒细胞或淋巴细胞性血管炎[7]。BD 中神经系统受累相对较少,发病率约2.3%~38.5%[5],约75%的神经系统受累患者有实质神经系统病变,表现为偏瘫、构音障碍、共济失调、颅神经病变和急性或亚急性发作的严重头痛。BD 累及胃肠道病变的主要症状是排便习惯改变、体质量减轻、腹痛、腹泻和胃肠道出血[8]。
2.2 BD 合并CDI 的發病机制
BD 合并CDI 具体发病机制尚不明确,但既往文献里其病机制的推测是以影像学研究进展为基础,且主要依据下丘脑、垂体的异常变化。2003 年的文献报道,Szymajda 等[9]认为其可能是BD 通过血管机制导致的CDI,说明患者脑部磁共振成像(magneticresonance imaging,MRI)示横窦血栓形成,垂体后叶缺乏高强度信号,其认为BD 会累及血管系统,导致血管损伤,出现毛细血管、小静脉、静脉及动脉炎。除了影像学研究外,Iwama 等[10]通过免疫学和分子技术发现rabphilin-3A是与CDI相关的活检证实的漏斗小管垂体炎患者的主要自身抗原,且仅在垂体后叶和下丘脑血管加压素神经元中表达,不在垂体前叶表达。本文报道的病例患者鞍区MRI 提示垂体后份未见确切T1WI 高信号神经垂体及垂体柄稍见可疑增粗,可能与BD 通过血管机制导致的CDI有关,但鞍区MRI 也提示垂体上份垂体柄旁见小斑片状T1WI 稍高信号,与正常垂体强化相仿,考虑可能是神经垂体异位,因此,BD 累及垂体后叶异常,不仅表现在垂体后叶无正常明亮信号,也可能表现为神经垂体异位。
2.3 BD 合并CDI 的诊断
最新的BD2014 年欧洲国际标准[11]:①眼部病变2 分;②生殖器溃疡2 分;③口腔溃疡2 分;④皮肤损害1 分;⑤神经系统损害1 分;⑥血管表现1分;⑦针刺反应阳性1 分,其中满足评分总和≥4 分即可诊断为BD。本例患者根据国际标准的②③项总分为4 分,故可诊断为BD。
本例患者无家族遗传史和外伤史,对于垂体前叶的小结节占位情况患者无明确的头痛、视力降低等占位性病变,且查各激素结果正常,因此排除家族遗传、创伤、占位性病变引起的CDI 可能。结合患者BD 病史,因此推测患者的CDI 可能是BD 此种自身免疫性疾病累及发展而来。但BD 合并CDI 目前仅国外有少数病例文献报道,并没有明确的诊断依据及病理学研究。不过有文献报道[12]自身免疫性尿崩症患者经蝶窦垂体活检可见垂体后叶和漏斗有大量浸润淋巴细胞和一些浆细胞,并在疾病发展后期出现纤维化[10],因此后续抗体检测可能更加需要完善。
2.4 BD 合并CDI 的治疗
根据本例患者具体病情使用了沙利度胺、甲氨蝶呤两种免疫抑制剂及甲泼尼龙激素治疗,辅以康复新液外用通利血脉、养阴生肌,可以有效缓解患者口腔、生殖器溃疡症状。对于CDI 的治疗,据报道,充足的补液、基础疾病的治疗和去氨加压素给药是治疗CDI 的主要手段[13]。而针对BD 合并的CDI,根据国外相关病例文献报道[9,14-15],给予去氨加压素治疗后尿崩症状均有好转。因此,针对本例患者的复杂病情表现,临床将BD 的免疫抑制、激素治疗和尿崩症的改善排尿治疗结合,能有效地控制和改善患者病情,但在CDI 急性脱水期时,应注意及时补液,避免引起高钠血症。
综上所述,通过对本例病例的报道及文献复习,旨在其发病机制和诊治提供新思路。
[参考文献]
[1] YAZICI Y, HATEMI G, BODAGHI B, et al. Beh?etsyndrome[J]. Nat Rev Dis Primers, 2021, 7(1): 67.
[2] MAHMOUDI M, ASLANI S, MEGURO A, et al. Acomprehensive overview on the genetics of Beh?et'sdisease[J]. Int Rev Immunol, 2022, 41(2): 84–106.
[3] TOMKINS M, LAWLESS S, MARTIN-GRACE J, et al.Diagnosis and management of central diabetes insipidusin adults[J]. J Clin Endocrinol Metab, 2022, 107(10):2701–2715.
[4] PATTI G, IBBA A, MORANA G, et al. Central diabetesinsipidus in children: Diagnosis and management[J].Best Pract Res Clin Endocrinol Metab, 2020, 34(5): 101440.
[5] DAVATCHI F, CHAMS-DAVATCHI C, SHAMS H, et al.Behcet's disease: Epidemiology, clinical manifestations, anddiagnosis[J]. Expert Rev Clin Immunol, 2017, 13(1): 57–65.
[6] FATEMI A, SHAHRAM F, AKHLAGHI M, et al.Prospective study of articular manifestations in Beh?et'sdisease: Five-year report[J]. Int J Rheum Dis, 2017,20(1): 97–102.
[7] WILLIAMS DAVID S. Behcet's disease[J]. J Insur Med,2019, 48(1): 103–105.
[8] SKEF W, HAMILTON M J, ARAYSSI T.Gastrointestinal Beh?et's disease: A review [J]. World JGastroenterol, 2015, 21(13): 3801–3812.
[9] SZYMAJDA A, ELEDRISI M, PATEL R, et al. Diabetesinsipidus as a consequence of neurologic involvement inBehcet's syndrome[J]. Endocr Pract, 2003, 9(1): 3–35.
[10] IWAMA S, SUGIMURA Y, KIYOTA A, et al.Rabphilin-3A as a targeted autoantigen in lymphocyticinfundibulo-neurohypophysitis[J]. J Clin EndocrinolMetab, 2015, 100(7): E946–954.
[11] BLAKE T, PICKUP L, CARRUTHERS D, et al.Birmingham Beh?et's service: Classification of diseaseand application of the 2014 international criteria forBeh?et's disease (ICBD) to a UK cohort[J]. BMCMusculoskelet Disord, 2017, 18(1): 101.
[12] SCHERBAUM W. Autoimmune diabetes insipidus[J].Handbook of Clinical Neurology, 2021, 181: 193–204.
[13] LEVY M, PRENTICE M, WASS J. Diabetes insipidus[J].BMJ (Clinical Research), 2019, 364: 1321.
[14] JIN-NO M, FUJII T, JIN-NO Y, et al. Central diabetesinsipidus with Beh?et's disease[J]. Intern Med, 1999,38(12): 995–999.
[15] GUMà A, AGUILERA C, ACEBES J, et al. Meningealinvolvement in Beh?et's disease: MRI[J]. Neuroradiology,1998, 40(8): 512–515.
(收稿日期:2022–08–16)
(修回日期:2023–02–18)
