全基因组关联分析中混合模型的原理、优化与应用
2023-06-27谭力治赵毅强
谭力治,赵毅强
全基因组关联分析中混合模型的原理、优化与应用

中国农业大学生物学院,北京 100193
全基因组关联分析(genome-wide association study,GWAS)是定位基因组中与性状显著关联的变异位点的有效方法。随着表型记录的完善、高通量基因型分型技术的发展,以及统计方法的改进,全基因组关联分析在人类疾病、动物植物遗传等领域得到了广泛的应用。假阳性是影响全基因组关联分析结果可靠性的重要因素之一。为了控制假阳性,除了校正值,GWAS模型从最简单的方差分析(或用于质量性状的卡方检验)到加入固定效应协变量的普通线性模型(general linear model,GLM),再到加入随机效应的混合线性模型(mixed linear model,MLM)持续改进,控制了多种混杂因素导致的假阳性。将个体的遗传效应拟合为由基因组亲缘关系矩阵(genomic relationships matrix,GRM)定义的随机效应是目前常用的方法。由于MLM的参数估计大量消耗计算资源,研究人员不断尝试模型求解优化和GRM的构建优化(GRM的构建优化同时也提高了计算效率),最终将基于MLM计算的时间复杂度由O(MN3)逐步改进到O(MN),实现了计算速度与统计功效的飞跃。针对质量性状病例对照比失衡带来的假阳性问题,研究人员进一步对广义混合线性模型(generalized linear mixed model,GLMM)进行了校正。本文较全面地介绍了GWAS的基本原理和发展,着重阐述了GWAS中MLM模型的改进和优化细节,同时,列举了GWAS在农业中的应用,包括在植物、动物和微生物方面的研究成果,以及基于单倍型的GWAS应用。最后,从进一步提高GWAS统计功效和GWAS试验设计2个角度对GWAS未来的发展进行了展望。
全基因组关联分析;复杂性状;随机效应;基因组亲缘关系矩阵;混合线性模型
1 概述
1.1 GWAS的基础
20世纪以来,作为一种有效的候选基因定位方法,连锁分析广泛用于定位孟德尔性状和常见疾病的基因和变异[1]。连锁分析在家系内寻找性状与标记等位基因的共分离,对于单基因性状的定位具有较高的精度。对于人类遗传病等复杂性状的遗传位点,由于单个变异的边际效应过小,其方法具有较大的局限[2]。人类基因组计划(human genome project,HGP)在2001年发表了人类基因组草图,成为基因组研究的一个重大进步。人类基因组单体型图计划(the international hapmap project,HapMap)于2002年启动,旨在建立人类全基因组遗传变异图谱。HapMap计划基于2个重要的遗传概念:其一是遗传变异和表型变异存在关联;其二是标记之间的连锁不平衡(linkage disequilibrium,LD)。在LD区间内,变异之间的信息冗余,LD的程度决定需要多少个遗传变异来对全基因组进行“标记”。这两点也成为全基因组关联分析(genome-wide association study,GWAS)重要的理论基础。
在人类基因组计划完成后的十年,以Illumina Infinium和Affymetrix原位光刻为代表的高密度芯片分型技术,以及以Solexa为代表的下一代测序技术(next generation sequencing,NGS)飞速发展,大大提高了基因分型的通量并降低了分型成本。众多物种基因组图谱的绘制和高通量基因型分型技术的进步,为GWAS研究提供了丰富的标记信息,促进了人类和动植物疾病以及复杂性状的遗传定位。另一方面,GWAS的统计模型从非参数卡方检验到普通线性模型(general linear model,GLM)再到混合线性模型(mixed linear model,MLM)持续改进,MLM成为当前GWAS的首选方法。与其他方法相比,MLM可以同时捕获由于群体分层、家系结构和潜在关联而产生的混杂效应,实现了更高的统计功效[2-4]。尽管与简单的模型相比,基于MLM的GWAS分析计算量非常大,但随着研究者的不断努力,目前大规模GWAS的计算已经被优化到人们可以接受的程度,其算法的时间复杂度实现了O(MN3)—O(MN2)—O(MN1.5)—O(MN)的巨大进步[5-7]。
1.2 GWAS的基本原理
尽管连锁不平衡也是GWAS的理论基础之一,GWAS并不依赖于家系而是利用群体的历史重组信息。常见疾病-常见变异假设(common disease common variant,CDCV)认为常见疾病由多个在群体中普遍存在的变异共同导致,每个变异都对疾病发生产生贡献。基于CDCV假设,GWAS使用丰富的单核苷酸多态(single nucleotide polymorphism,SNP)为标记,扫描表型在整个基因组中的关联信号。
GWAS中各位点通常独立进行检验。对于二元质量性状,数据可以表示为一个列联表,表中每个元素为具有特定基因型-表型组合类别的个体数量,使用卡方检验判断类别之间的独立性。遗传模型的类型决定了列表的形式,例如,在显性或隐性模型的情况下是一个2×2的列联表,而加性模型则用一个2×3的列联表表示。由于加性模型认为基因型与表型具有有序关系,在趋势或有序的假设下,也可以使用Cochran- Armitage趋势检验来捕捉这种关系。
在实际应用中,研究人员可能希望加入协变量来控制混杂因素,例如,某种疾病的患病概率往往随着年龄的增长而增加,或者与性别相关。使用线性模型可以方便地将年龄和性别作为协变量加入,对模型进行调整。对于质量性状,最常用的线性模型是logistic回归模型[8]。而对于数量性状,则使用GLM。
群体分层是另一类主要的混杂因素。群体分层指亚群体间等位基因频率的系统性差异。从群体遗传学的角度看,群体分层可能由于选择压力或者遗传漂变所致。群体分层可能造成基因型和表型的虚假关联。如图1所示,子群体1和子群体2之间存在明显的等位基因频率差异,单独来看,2个子群体的OR值(odds ratio)均为1。将2个子群体结合后,合并后群体的OR值达到2.87,提示群体分层造成了假阳性结果。为了识别真正的关联信号,控制群体分层很有必要[9]。
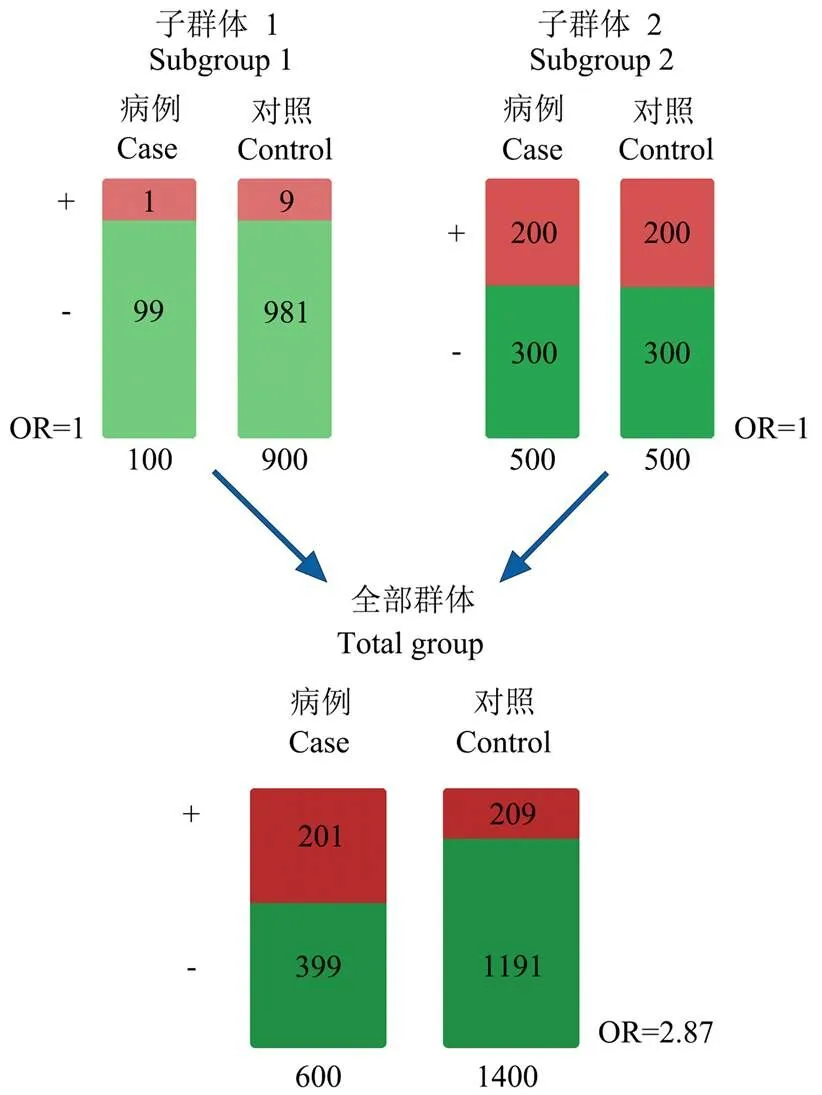
图1 群体分层示例
控制群体分层的第一种方法是使用单一群体确保群体同质性。第二种方法是基于家系的设计,从直系子女和全同胞个体中挑选不同表型的个体进行分析。第三种方法是使用Devlin等[10]提出的基因组控制方法,使用Cochran-Armitage趋势检验来计算膨胀系数,用来校正卡方统计量的膨胀。然而这些方法存在一些局限性,比如难以获得足够符合要求的样本,方法的适用性有限,以及使用统一的调整忽略了个体间基因型的差异。为了解决这些局限,人们开发了另一种替代方法,即用基因型主成分来捕捉群体结构[11]。主成分分析抽取数据中的主要变异,被证明能够准确反映种群之间整体的遗传差异,人们将基因型前几个主成分用作协变量来校正群体分层产生的影响。由于其计算简单,效果较好,基于主成分的校正是目前最常用的控制群体分层的方法。
由于目前的全基因组分型芯片或全基因组测序提供的基因座数量可达上百万,因此,GWAS中对每个基因座单独检验后的显著性校正尤为重要。Bonferroni校正是多重检验校正的经典方法,然而,使用Bonferroni校正GWAS阈值可能由于LD的存在而变得过于保守[12]。人中GWAS普遍使用的阈值为5×10-8,该阈值基于假设独立的SNP数目大约为100万个,其阈值随SNP数目的增加而逐渐严格。相比于Bonferroni校正,FDR(false discovery rate)校正是一种“相对温和”的校正方法。该方法不追求完全避免假阳性的结果,而是将假阳性结果和真阳性的比例控制在一定范围内,一般选择信号的前5%作为FDR校正的标准。此外,使用置换检验(permutation test)来获得调整后的显著性值被认为是最好的校正方法,但这种方法的计算量大,尽管使用其近似方法[13]可以提高计算效率,但在实际应用中几乎仍是不可行的。
2 混合线性模型在GWAS中的发展
2.1 从普通线性模型到混合线性模型
在线性模型中,通常使用样本来自亚群的比例或全部基因型计算所得的主成分(principal component,PC)表示群体结构,称其为Q矩阵[10, 14]。Q矩阵中的协变量被用作固定效应进行拟合。该模型表示为=++,其中,和分别是表型和单个遗传标记(SNP),是残差。这个GLM也被称为Q模型。
除了群体分层,人们意识到来自个体的效应(或等价的多基因效应)也是产生结果偏倚的因素。个体效应的自相关结构可以通过个体之间的亲缘关系矩阵来指示[15],早期在动物中多使用基于系谱的亲缘关系矩阵,由于植物系谱往往未知,因此,无法将在动物中使用的策略直接用于植物[16]。而使用全基因组遗传标记可方便地计算基因组亲缘关系矩阵(genomic relationships matrix,GRM)(此处也称为K),此时个体的遗传效应被拟合为由K定义的随机效应。同时具有Q和K的模型是MLM,表示为=+++,也称为Q+K模型[4]。研究表明,Q和Q+K模型较好地控制了假阳性,而且Q+K模型比Q模型或单独的K模型表现更好[4, 15]。
在最初的MLM模型中[4],Q+K方法的混合模型方程表示为:
=++++
式中,为表型向量;是除SNP或种群结构以外的固定效应;是SNP固定效应;是群体结构固定效应;是多基因随机效应;是残差;、、、是分别与、、、对应的设计矩阵。随机效应的方差()=KV,是个体亲缘关系n阶方阵,V为多基因遗传方差;表型的方差=KV+RV,是n阶单位矩阵,V为残差方阵。通过求解混合模型方程,可获得、、(固定效应)的最佳线性无偏估计值(best linear unbiased estimate,BLUE)和(随机效应)的最佳线性无偏预测值(best linear unbiased prediction,BLUP)。MLM进一步避免了个体相关导致的假阳性结果[17]。
2.2 GWAS中混合线性模型算法的优化
最大似然法(maximum likelihood,ML)或约束最大似然法(restricted maximum likelihood,REML)常用于MLM的方差组分估计。与GLM相比,MLM的计算量非常庞大,研究人员从不同角度对MLM用于GWAS进行计算效率和统计功效上的优化,表1总结了GWAS中MLM的优化模型。Kang等[18]提出了高效混合模型关联(efficient mixed model association,EMMA)法。在似然估计中,EMMA通过把遗传方差和残差方差2个组分的优化简化为对两者比值的优化,并通过特征分解简化参数估计中的迭代运算,显著提高了MLM的求解速度。
GWAS中使用成百上千的个体和成千上万的标记,对每个标记进行检测时都估计一次随机效应的方差使得全基因组分析效率低下。Kang等[19]又提出了改进的高效混合模型关联(efficient mixed-model association expedited,EMMAX)法,该方法认为由位点多基因效应捕获的个体随机效应在模型中的贡献很小。基于这样的假设,EMMAX法把对随机效应方差的多次估计改为单次估计并将其在模型中固定。EMMAX法可以视作EMMA法的简化近似方法,在随机效应方差组分估计完成后,采用GLM单独估计每个标记的效应,计算速度相对于EMMA获得了大幅提升。
压缩MLM法(compressed mixed linear model,CMLM)[20]通过聚类把相近个体划分为不同组别,通过组间GRM来替代个体间GRM实现对随机效应的压缩。由于随机效应的计算和个体数的三次方成正比,此方法大大节省了计算时间。Li等[21]对CMLM算法进行改进,从8种聚类算法与3种组间亲缘关系算法的24种组合中计算最优组合,称其为增强CMLM(enriched CMLM,ECMLM),把CMLM的检测功效提高了10%左右。在提出CMLM的同时,Zhang等[20]也提出了提前确定模型的群体参数(population parameters previously determined,P3D)的两步法优化策略,第一步通过没有标记效应的简化模型估计总的遗传方差、残差以及聚类数等群体参数,这些参数作为先验信息在第二步的模型中固定使用。第二步依然使用MLM,把原始表型和已估计的参数用于模型中估计标记效应。CMLM和P3D可单独或联合使用来优化计算和提供统计功效,相对于常规MLM获得数千倍的效率提升。
Lippert等[5]提出因式谱变换线性混合模型(factored spectrally transformed linear mixed models,Fast-LMM)。该方法的核心是对GRM进行一次特征分解,转化为多个不相关的矩阵后使其能使用GLM进行高效求解。由于方法并不要求每个标记具有相同的效应,Fast-LMM方法得到的是各标记效应的精确估计。作者将Fast-LMM与EMMAX进行比较,在(wellcome trust case control consortium,WTCCC)克罗恩病(Crohn’s disease)数据集的4 000与8 000个标记中,Fast-LMM的计算速度分别为EMMAX的11和5倍。
Aulchenko等[22]首次提出了简单快速的GRAMMAR法(genome wide rapid association using mixed model and regression)进行GWAS。GRAMMAR分为两步:第一步使用GRM作为随机效应和除标记效应外的协变量做固定效应建模。第一步模型的残差作为第二步模型的表型,第二步使用GLM仅对每个标记单独建模。GRAMMAR法第一步中MLM相对耗时,而剥离了随机效应的第二步GLM非常高效。为了改进GRAMMAR在标记效应估计上的偏差,作者团队提出了改进的二步法GRAMMAR-Gamma[23]。该方法在第一步中通过特征分解加速矩阵运算,并构造了不考虑标记相关性的简化得分检验(score test)统计量和GRAMMAR-Gamma校正因子。在第二步中仍然使用GLM对每个标记单独建模,但对检验统计量除以GRAMMAR-Gamma因子进行校正以获得更精确的标记效应的估计。该方法取得了与标准似然检验几乎相同的功效,但极大降低了运算时间。作者使用人类和拟南芥的数据发现GRAMMAR-Gamma的运行时间远小于EMMAX和Fast-LMM,在运行速度上分别为二者的38和10倍,再次提高了计算效率。
全基因组高效混合模型关联(genome-wide efficient mixed-model association,GEMMA)法[24]在对GRM进行特征分解之前,对矩阵求一阶和二阶导数。为了避免EMMA中对每一个标记复杂的特征分解步骤,使用矩阵向量乘法替代特征分解,转化为只涉及标量的递归乘法优化求解。GEMMA法通过优化大型矩阵运算,显著提高了GWAS的运行速度,并获得和EMMA法一致的精确解。使用杂交小鼠的高密度脂蛋白胆固醇(high-density lipoprotein cholesterol,HDL-C)数据,作者报道GEMMA的运行速度为Fast-LMM的13.6倍,而使用WTCCC的克罗恩病数据,GEMMA的运行速度为Fast-LMM的1.87倍。作者同时指出,EMMAX与GRAMMAR等近似方法可能导致假阴性,造成GWAS检测的功效降低。
Fast-LMM的作者进一步提出使用全部标记的子集构建GRM来降低计算开销,称为FaST-LMM- Select[25]。在操作中,作者首先使用GLM对标记进行检验并对值进行升序排序。选取达到基因组控制因子λ时对应的标记集合,剔除其中强连锁的标记,使用剩余标记构建性状特异的GRM。该方法进一步降低了计算成本,且作者发现相较于使用全部标记,该方法能显著降低假阳性和假阴性率。Wang等[26]提出的SUPER(settlement of mlm under progressively exclusive relationship)方法运用了类似的概念来优化GRM的构建。该方法对标记预先获得的值或效应值进行排序,在划分好的若干染色体片段中,选取每个片段中值最低的标记并剔除与待测标记连锁的标记后,同样使用剩余标记构建性状特异的GRM。经作者比较,该方法比FaST-LMM-Select的假阳性更低,并提高了对遗传力的估计。之后发表的BOLT-LMM[6]法包括两部分,基础部分和主流方法一样采用微效多基因假设下的混合线性模型。但是在估计方差组分时使用共轭梯度法实现近似计算,避免了特征分解所需的大量计算时间和内存。作者采用一种新的回顾性得分检验并使用类似GRAMMAR-Gamma的方法对统计量进行校正。BOLT-LMM的改进部分借鉴了贝叶斯方法在动植育种基因组选择中的应用,认为大部分标记效应较小但存在少部分大效应标记,其假设更贴合实际。作者使用高斯混合分布作为标记效应的先验分布来拟合贝叶斯线性回归,通过快速变分法得到近似的表型残差。最后,基于表型残差使用同样的回顾性得分对每个标记进行检验并使用LD分数回归(LD score regression,LDSC)[27]对统计量进行调整。使用23 294个人样本的脂质、身高、体重指数和血压等定量性状,作者证明BOLT-LMM具有更高的统计功效,且运行效率相较FaST-LMM-Select、GEMMA和EMMAX等更高。
Jiang等[7]开发了基于MLM的GWAS工具fastGWA,采用高效的基于网格搜索的约束性最大似然(restricted maximum likelihood,REML)算法fastGWA-REML。在对GRM稀疏化的基础上,方差组分估计时对矩阵使用Cholesky分解避免对其求逆。fastGWA使用了与GRAMMAR-Gamma类似的统计量检验关联性。fastGWA方法比其他基于MLM的工具快几个数量级,其内存使用量也极大降低。作者抽取了英国生物样本库(UK Biobank,UKB)中400 000个样本,分别使用fastGWA与BOLT-LMM对体重指数进行GWAS分析。fastGWA的运行时间为BOLT-LMM的1.22%,内存使用量仅为BOLT-LMM的5%,使得Biobank级的GWAS运算成为可能。
上述方法更多聚焦于MLM,尤其是针对模型中作为随机效应的GRM的计算优化,个别方法在构建GRM时做了一些简化,但是总体思路接近,方法的统计功效依然与传统的MLM类似。MLM较好地控制了假阳性,但是人们意识到,在一定程度上其存在标记效应和控制效应的混杂,造成了一定程度的假阴性结果。于是一些新的方法尝试在模型中剥离随机效应,从而解决这类混杂问题。
多位点混合模型(multi-locus mixed-model,MLMM)法[28]采用前向后向选择组合的逐步选择策略对常规的MLM进行改进。在前向选择中,每一步都首先估计方差组分,将最显著的标记作为固定效应的协变量逐步加入模型,用来压缩模型中的随机效应。过程中持续更新模型的方差组分,直到随机效应解释的变异接近于零或达到指定的循环数后结束前向选择。类似地,在随后的后向选择中逐步把最不显著的标记协变量从模型中剔除。为了避免近端污染(proximal contamination),作者建议模型中作为协变量的标记不参与GRM的构建。
MLMM法使用向前向后逐步回归消除了一部分检测标记效应的混杂问题,增强了GWAS的统计功效。在此基础上,Liu等[14]提出交替使用固定效应和随机效应(fixed and random model circulating probability unification,FarmCPU)来进一步解决模型中混杂问题的方法。对每一个标记,该方法把筛选后可能的关联位点作为协变量加入固定效应模型来进行检测。而候选关联位点的筛选是在独立的随机效应模型中进行,更新候选关联位点后重新进入固定效应模型对标记进行检测,循环往复直到没有新的候选关联位点加入到固定效应模型中。由于模型中没有同时出现固定和随机效应,FarmCPU方法避免了不同效应的混杂,并同时控制了假阳性和假阴性。作者团队针对FarmCPU的改进方法BLINK(Bayesian-information and linkage-disequilibrium iteratively nested keyway)法[29]更是不再使用随机效应模型,而是使用连锁不平衡和贝叶斯信息准则(Bayesian information criterion,BIC)来入选和筛选可能的关联位点。作者使用模拟数据对BLINK和FarmCPU进行评估,发现BLINK的运行速度是FarmCPU的3—4倍,且BLINK更具有发掘额外遗传位点的潜力。本文在单个处理器上(Intel(R) Xeon(R) Gold 6230 CPU @ 2.10GHz)比较了上述模型的单核运算时间。从Galbase数据库[30]下载928个样本的基因型数据,随机抽取GGA 11上443 218个位点用于GWAS测试。表型数据使用GCTA[31]进行模拟,选择100、200、400、600、800和928个样本进行不同样本量梯度的测试。经测试,EMMA、CMLM、SUPER运算时间依次减少,但远高于其他模型(图2)。FastGWA运算速度最快,其次为GEMMA、BLINK、FarmCPU,其运算时间在样本量较大时明显低于其他模型。BOLT-LMM、MLMM、FaST-LMM、EMMAX运算时间高于上述4个模型,可能由于样本量和标记数目的限制,未能观察到明显差距(图2)。

表1 GWAS中MLM的优化模型

图2 混合模型算法概述与运算速度比较
2.3 GWAS中的广义混合线性模型算法
随着许多大型生物样本库和队列的建立,基于MLM的GWAS在数量性状中取得了巨大的成功。然而大部分人类遗传病是病例对照(case-control)研究,属于二分类的质量性状,患病率较低的疾病还可能出现病例-对照比极不平衡的现象,这样的数据不满足MLM中残差具有方差均等的假设。当存在群体分层,尤其病例-对照比不平衡时,基于MLM的GWAS可能无法有效地控制Ⅰ型错误率(假阳性),因此,研究者们对MLM做出扩展,将更适合此类情况的广义混合线性模型(generalized linear mixed model,GLMM)应用于GWAS中,表2汇总了GWAS中GLMM的优化模型。
Chen等[32]提出广义线性混合模型关联检验(generalized linear mixed model association test,GMMAT),该方法基于残差异方差的logistic混合模型。方法首先构建不含标记效应的零模型,模型拟合中作者使用惩罚拟似然法(penalized quasi-likelihood,PQL)和平均信息约束最大似然法(average information restricted maximum likelihood,AI-REML)进行参数估计。估计得到的模型参数被固定下来用于所有标记,并在此基础上使用得分检验对每个标记效应进行估计。作者比较了GMMAT和SAS的PROC GLIMMIX过程,发现GMMAT在拟合具有一个方差分量的logistic混合模型时,其运算时间为SAS PROC GLIMMIX的1.5%。此外,在存在不平衡的病例-对照比的情况下,GMMAT更好地控制了假阳性。
Zhou等[33]提出了可扩展的精确广义混合模型(scalable and accurate implementation of generalized mixed model,SAIGE)法,用于处理病例对照比失衡的大规模质量性状数据。SAIGE法的步骤与GMMAT类似,但是过程中大量使用了计算优化。SAIGE法第一步使用AI-REML估计方差组分等参数,使用预处理共轭梯度法(preconditioned conjugate gradient,PCG)替代矩阵特征分解,节省了GRM相关的计算成本。第二步使用方差比来校准得分统计量的方差,并借助鞍点近似法(saddlepoint approximation,SPA)克服二分类性状中病例对照比失衡的问题,获得准确度高的值。作者从UKB中随机抽取了冠状动脉疾病样本,分别使用BOLT-LMM、GMMAT和SAIGE进行GWAS分析。结果表明,SAIGE的假阳性率显著优于前两者。SAIGE的时间复杂度与BOLT-LMM相同,但由于logistic混合模型的迭代步骤多于MLM,导致其运算速度低于BOLT-LMM,但依然快于GMMAT。
同样使用SPA来校正病例对照比失衡带来的假阳性问题,Jiang等[34]开发的fastGWA-GLMM工具沿用了前序fastGWA法中基于网格搜索的算法估计方差组分,并使用GRM稀疏化提高计算效率。作者通过从UKB中随机抽取样本,将fastGWA-GLMM与SAIGE进行比较,在样本量为400 000时,fastGWA-GLMM的运行速度为SAIGE的36.8倍,极大地节省了运算成本。
除病例-对照研究产生的二分类质量性状外,临床上常使用有序分类测量来衡量疾病的严重程度,如从1—9对疾病的感染程度打分,1为基本不感染,9为严重感染。把有序分类变量当作连续变量或者降级为二分类变量使用都不合适,基于此,Bi等[35]提出了比例优势logistic混合模型(proportional odds logistic mixed model,POLMM)法。该方法使用和上述方法相似的统计架构,将logistic模型应用于有序分类表型,使用PQL与AI-REML对零模型进行拟合并进行参数估计,也通过SPA校准第二步得分检验的值。根据使用的GRM的不同,POLMM法提供DensePOLMM与FastPOLMM 2种实现,DensePOLMM使用稠密矩阵并通过PCG进行矩阵加速运算,FastPOLMM使用稀疏矩阵,在计算速度上有优势但统计功效略低于DensePOLMM。作者使用BOLT- LMM、FastPOLMM-NoSPA和FastPOLMM对UKB中4种食品偏好的有序分类变量进行GWAS。当表型分布平衡时,BOLT-LMM获得与FastPOLMM一致的结果;当表型分布不平衡时,FastPOLMM-NoSPA优于BOLT-LMM,而FastPOLMM较前两者更好地控制了假阳性。
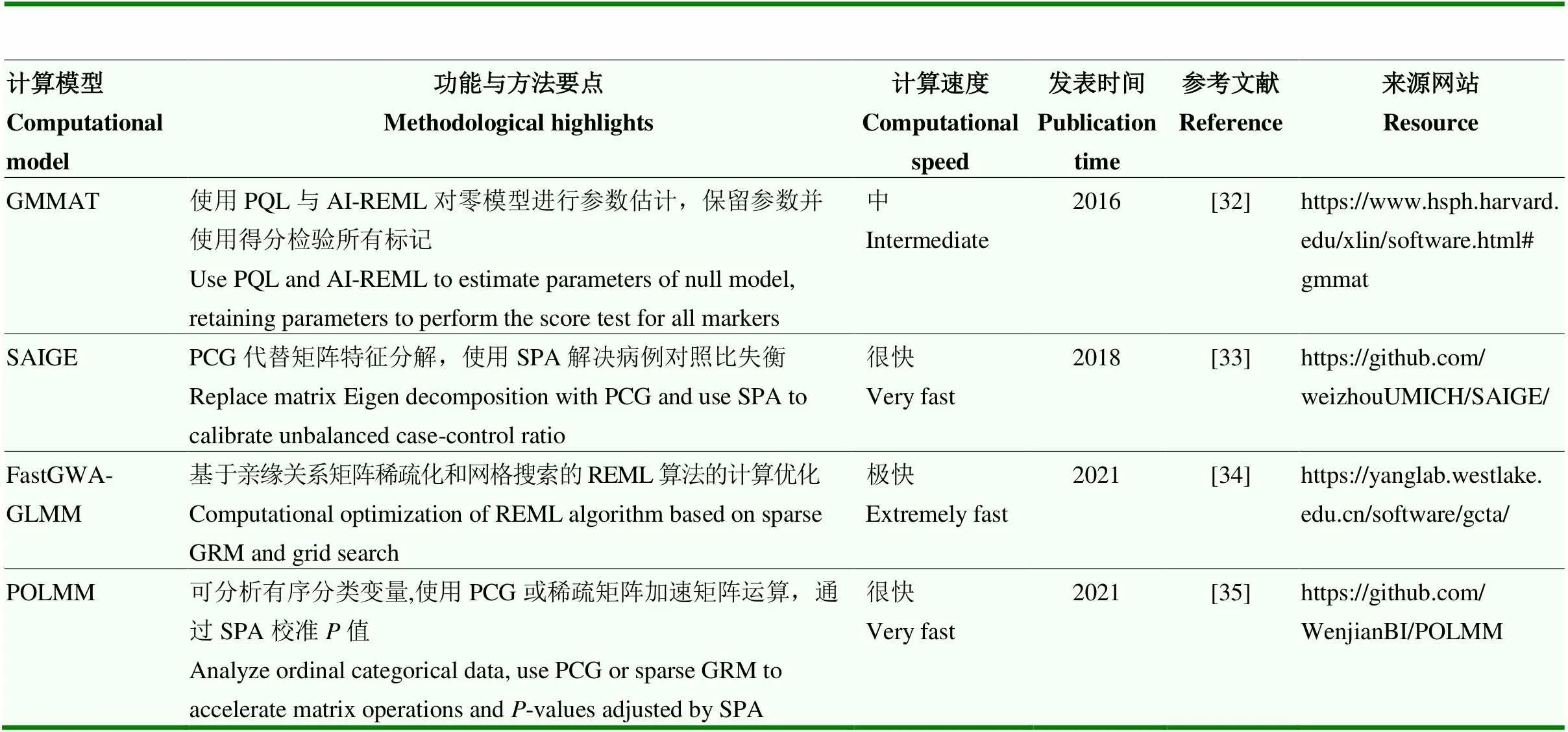
表2 GWAS中GLMM的优化模型
使用上述数据在单个处理器上(Intel(R) Xeon(R) CPU E3-1245 v6 @ 3.70GHz)比较不同样本量下GLMM模型的单核运算时间。与MLM测试相似,fastGWA-GLMM具有最快的运算速度,FastPOLMM几乎与其相同,SAIGE在样本量较小时速度较快,在样本量大于800时速度显著降低,DensePOLMM在4种模型中使用了最多的计算时间(图2)。
3 GWAS的应用
随着MLM在GWAS中的引入,越来越多和动植物重要性状关联的基因和位点被发现,推动了遗传学的发展。
3.1 GWAS在植物中的应用
自从Hansen等[36]使用BSA分析(bulked segregant analysis)首次将GWAS用于海甜菜后,GWAS方法已成功用于植物育种、逆境生长、抵御病虫害等方面的候选基因鉴定。Atwell等[37]使用EMMA对拟南芥自交系107种表型进行了GWAS分析,鉴定到6种受单基因控制的表型,包括抗病响应、开花基因表达等。
水稻与人类生活息息相关,Zhao等[38]对不同国家的413种水稻品种开展GWAS分析,利用EMMA鉴定到34个与植物生理、生长发育以及植物形态发生相关的候选基因。Huang等[39]采用低深度测序数据进行基因分型,使用CMLM法将GWAS应用于籼稻的14种育种相关性状,共筛选出37个与开花日期和增产性状显著相关的QTL,该分析促进了水稻优良农艺性状的遗传解析以及品种的选育。在全球950个水稻品种中,Huang等[40]使用CMLM分别对其中的地方粳稻群体、地方籼稻群体与全部品种进行GWAS分析,定位到32个与开花日期和农艺性状显著相关的QTL。产量是备受关注的农业性状之一,研究人员在利用TASSEL开展的基于混合线性模型的14种高产水稻品种的GWAS分析中,找到1 152个和高产相关的显著位点,同时检测到8个高产相关渗入区段,揭示了高产相关基因由于受到人工驯化而发生了遗传结构的改变[41]。
作为另一种重要的农作物,玉米中的GWAS研究也已广泛开展。Wang等[42]通过TASSEL软件在114种玉米自交系品种中定位到18个与抗黑穗病显著相关的变异位点,同时揭示了玉米抗黑穗病的遗传机理。Li等[43]利用CMLM在368种玉米自交系的103万个变异位点中鉴定出74个与籽粒油分和脂肪酸形成相关的候选基因,结合eQTL(expression QTL)与共表达分析发现有三分之一的候选基因与油分的代谢途径相关。同样,研究人员使用CMLM进行GWAS分析,发现玉米的早花性状与220个遗传标记显著关联,其中大部分关联位点与拟南芥同源[44]。绿色保持是植物延缓衰老的一种表现形式,Sekhon等[45]通过测定叶绿素荧光参数v/m指示植物延缓衰老的程度,使用GAPIT软件进行关联分析,鉴定到64个与其显著相关的候选基因,其中14个基因已被证明与衰老过程相关。作者同时构建了延缓衰老与正常衰老基因的共表达网络,对候选基因的生物学功能作出系统解释。Chao等[46]通过TASSEL软件对玉米内核锌浓度进行GWAS分析,鉴定到锌-烟酰胺转运蛋白基因,并验证了该基因过表达能够使玉米粒中锌浓度增加31.6%,实现玉米中锌的生物强化。
3.2 GWAS在动物中的应用
GWAS也是动物遗传分析的主要研究方法之一,通过定位影响家畜重要经济性状的变异位点和候选基因,帮助研究者更好地理解性状的遗传机制,在家畜育种和改良方面起到重要的推动作用。
在1 027头杜洛克和二花脸杂交的F2代群体的GWAS分析中,Ren等[47]发掘出多个与复杂性状显著相关的候选基因:使用SAS的GLM模型发现猪耳朵大小受到PPARD进化保守区域变异的影响;使用R包GenABEL发现在糖原分解的级联激活中发挥作用,其突变导致骨骼肌糖酵解潜能增加,进而影响猪肉产量[48]。Wang等[49]在82头母猪中使用CMLM进行GWAS分析,鉴定到266个与猪出生重显著相关的QTL。Guo等[50]分别使用MLM与贝叶斯混合模型,共定位到15个与仔猪数目与死亡率性状有关的候选基因。Gozalo-Marcilla等[51]使用GEMMA对来自8个具有不同遗传背景的275 590头猪的背腩厚度性状进行GWAS分析,共定位到264个位点与背腩厚度显著关联,鉴定了64个与脂肪代谢相关的候选基因。
鸡的GWAS分析大多聚焦于生长、产蛋与抗病性状。Gu等[52]使用乌骨鸡与白洛克鸡杂交的F2群体,利用PLINK软件定位到基因组中3个与鸡体重相关的区段。该课题组后续进行了多个种鸡不同表型性状的GWAS研究,包括羽毛形态、胡须与鸡冠形态等方面[53-54],同时使用祖先单倍型对鸡的生长性状进行精细定位,筛选出9个关键的候选区段供进一步研究[55]。在400只中国鸡的生殖性状GWAS分析中,Fan等[56]利用PLINK软件鉴定到19个与蛋重、产蛋数等性状相关的QTL,定位到、、等17个候选基因。Li等[57]使用GEMMA对839只母鸡的多个蛋壳晶体结构相关性状进行GWAS分析,鉴定到GGA1上55.6—69.1 Mb区间内部621个显著信号,注释到、、、、和等参与调节胞质钙离子浓度生物过程的基因。Guo等[58]通过建立肉鸡外翻-内翻畸形(valgus-varus deformity,VVD)病例组与对照组,基于233个样本使用GEMMA进行GWAS分析,筛选到5个与VVD显著相关的变异,并将GWAS与转录组分析整合,定位到重要的易感基因。
羊是中国重要的农业动物,其种类繁多,且具有丰富的遗传资源[59]。Demars等[60]使用PLINK对2种羔羊开展GWAS后定位到与高产表型和排卵率显著关联,揭示了在羔羊卵巢发育功能中的关键作用,为探索生育障碍提供了重要依据。He等[61]将GWAS分析用于3个不同品种的中国本土绵羊,使用CMLM在双角羊和四角羊中鉴定到和2个候选基因,帮助理解绵羊角发育的分子调控。
水产养殖的遗传收益总体上高于陆地农业动物[62],水产的抗病育种具有重要的经济意义。大西洋鲑鱼普遍受到细菌性肾病的侵扰,Holborn等[63]使用GenABEL对507只大西洋鲑鱼对细菌性肾病下的抗性进行GWAS分析,发现其抗性为多基因性状,并定位到2个与细菌性肾病抗性显著相关的QTL。Peng等[64]使用PLINK对黄河鲤鱼体重、体长和胴体重量等性状进行QTL定位,鉴定出多个与神经发育,基础代谢相关的基因,为黄河鲤鱼生长性状的选育提供了遗传材料。黄花鱼同样是我国的传统养殖鱼类之一,黄花鱼具有明显的性别二态性,雌性黄花鱼的生长性状显著优于雄性。LIN等[65]对905只黄花鱼(463只雌性,442只雄性)开展GWAS分析,使用TASSEL在第21染色体处发现22个QTL与性别决定显著相关,鉴定到包括调控精子发育、雌性激素代谢功能的14个候选基因。同时,对不同性别性腺指数的GWAS确定了第18染色体与雄性性腺发育相关的区段,鉴定到相关调节基因、和。
3.3 GWAS在微生物中的应用
微生物与植物的整个生长过程息息相关。植物病原微生物在定植后导致植物产生特定疾病,从而影响植物生长。Davila等[66]使用Fast-LMM在350株拟南芥中鉴定出干旱条件下与灰霉菌()真菌病原体相关的转录因子,其与耐寒及坏死真菌抵抗相关。Zhang等[67]使用701种不同水稻种质和23种不同水稻白叶枯病菌(pv,)菌株进行跨物种GWAS,使用EMMAX筛选出47个毒力相关基因和318个水稻不完全抗性基因,并对毒力相关基因与抗病基因之间的基因互作加以阐述。Martins等[68]使用BLINK对豌豆的派伦霉菌()抗性以及茎直径等生长性状进行GWAS,发现具有抗病性的等位基因导致了较低的株高,印证了植物抗病以牺牲自身生长为代价这一结论[69]。de Ronne等[70]同时使用ECMLM、FarmCPU和BLINK在357个大豆品种中发现了新的大豆疫霉菌()抗性QTL,该QTL的LD区块内包含与病原体抗性相关的乳胶蛋白编码基因。
与病原微生物不同,根际微生物中存在一部分能够促进植物营养吸收的共生体[71]。根际微生物正向促进了植物的营养吸收能力与范围,同时调节植物的生长发育与抗性反应[72]。Bergelson等[73]使用EMMAX基于拟南芥的细菌丰度数据鉴定到与,其分别调控植物免疫与侧根形成;通过真菌的丰度数据发现SNARE蛋白的靶点,其参与根毛蛋白质转运。研究发现细菌与真菌丰富度的GWAS结果基本没有重叠,表明细菌与真菌群落丰富度受到不同基因的影响。Deng等[74]使用GEMMA研究了高粱遗传位点与根际微生物丰度的关系,并使用高粱遗传信息成功预测根际微生物组成情况。
肠道益生菌能够提高动物的饲料转化率,增加动物体重,以及增产牛奶或鸡蛋等农业产品,实现更多经济价值[75-78]。Crespo-Piazuelo等[79]使用GEMMA对285只猪肠道微生物中18个属的相对丰度进行GWAS分析,发现、、、、和与基因型存在显著关联,定位到包括免疫应答与生理调节相关的多个候选基因。Bergamaschi等[80]在1 028头猪在断奶期、生长中期、生长末期3个生长过程的粪便中提取微生物样本,提取肠道微生物Alpha多样性与分类操作单元(operational taxonomic unit,OTU)作为表型数据,使用EMMAX鉴定到候选基因。该基因在肠道组织中高度表达,与细胞增殖相关。
3.4 基于单倍型的GWAS应用
复杂性状GWAS研究有助于了解复杂性状的遗传机制,定位到的候选基因为进一步的研究提供了指导方向。单倍型是染色体上共同遗传的多个等位基因的组合,包含了等位基因间的连锁信息。单位点GWAS每次检验一个SNP,单倍型GWAS(haplotype- based genome-wide association study,hGWAS)把整个单倍型区块用于GWAS分析,检测与性状显著关联的单倍型区块。由于单倍型可能包含了位点之间的互作信息,一些发表的基于单倍型的GWAS分析证明hGWAS在定位效果以及统计意义上均强于基于单位点的GWAS[81-83]。
在水稻中,Yano等[84]把单倍型作为固定效应进行GWAS分析,筛选出4个与水稻农艺性状相关的候选基因。Ogawa等[85]使用日本的8个高产水稻品种构建了日本-多亲高代杂交系(Japan-multi-parent advanced generation inter-cross,JAM),通过8个祖先群体进行hGWAS分析,鉴定到控制糯性胚乳和糯素长度性状的QTL。与上述方法不同,Zhang等[86]开发了GLASCOW软件将祖先单倍型加入GLMM作为第二个随机效应组分,即GRM控制多基因效应而祖先单倍型控制群体分层,在3种单基因隐性疾病中获得了EMMAX未检测到的显著结果。在瘦鸡和肥鸡两个肉鸡品种的GWAS分析中,Zhang等[87]定位到与腹部脂肪重量显著关联的132个单倍型区块,筛选出7个可能在控制腹部脂肪含量中产生影响的候选基因。Howard等[88]在18 773个苏格兰家庭中发现了2种与重性抑郁障碍显著关联的单倍型,其中包括与双相情感障碍相关的单倍型区域,该结果通过25 035个UKB中的个体加以验证,为揭示重度抑郁症的遗传机制提供了思路。
4 展望
从MLM被引入GWAS起,研究人员持续对其进行优化。目前MLM在GWAS中的优化主要包括2种:其一为对随机效应求解中GRM相关的计算优化;其二为对随机效应中GRM构建的优化。方法的优化大幅控制了计算结果中的假阳性,并显著提升了计算速度。对于计算优化部分,fastGWA作为目前最快的MLM算法实现,已经将GWAS中混合线性模型的时间复杂度降低至O(MN),使大规模数据的快速GWAS计算成为现实。伴随着GWAS的广泛使用,累积了越来越多的汇总数据(summary statistics),具有高统计功效和计算速度的META-GWAS-MLM算法有待开发以利用这些汇总数据,实现GWAS的联合分析。另一方面,MLM控制了假阳性却带来了假阴性问题,降低了统计功效。近期发表的方法FarmCPU与BLINK尝试剥离随机效应,以解决MLM导致的假阴性问题。提高关联分析的统计功效可以考虑多种方式,使用单倍型的hGWAS可能是提高功效的方法之一。单倍型包含了可能的标记互作信息,比单位点信息量更丰富。在GWAS中引入贝叶斯思想同样能够增加GWAS的统计功效,因此,MLM与先验信息的结合也可能是未来控制假阴性的策略之一。协变量的优化同样可能是混合模型GWAS的优化内容之一,更好地捕获复杂数据全局和局部信息能更好地应对群体分层。对于复杂性状而言,基因多效性和多基因效应是影响表型形成的关键因素,目前这方面的研究相对较少。开发和完善多位点混合模型GWAS的方法对于遗传定位,以及研究复杂性状的形成机制具有重要作用。
基于混合模型的GWAS已经广泛应用于植物、动物和微生物的遗传研究,为生物育种提供了理论基础和新的思路。精心设计的GWAS试验有助于更精确地定位到候选基因,比如使用重组自交系(recombinant inbred line,RIL)或深度杂交系(advanced intercross line,AIL)的实验群体能够更好地定位候选基因。除此之外,基因与环境互作影响动植物优质性状形成的分子机制解析是未来GWAS研究的一个重要方向。目前已经广泛认识到环境对于植物生长性状具有重要的影响,适宜的环境可以显著增加作物产量。通过挖掘并改良作物的环境互作基因,以及表型可塑性的相关基因,有助于了解作物的育种潜力,并帮助作物在适宜环境中增加产量,在恶劣环境中维持产量,在育种中有着重要作用。而这些在动物的环境适应性研究中同样适用。
[1] BOTSTEIN D, RISCH N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nature Genetics, 2003, 33(3): 228-237.
[2] VISSCHER P M, BROWN M A, MCCARTHY M I, YANG J. Five years of GWAS discovery. The American Journal of Human Genetics, 2012, 90(1): 7-24.
[3] VISSCHER P M, WRAY N R, ZHANG Q, SKLAR P, MCCARTHY M I, BROWN M A, YANG J. 10 years of GWAS discovery: Biology, function, and translation. The American Journal of Human Genetics, 2017, 101(1): 5-22.
[4] YU J, PRESSOIR G, BRIGGS W H, BI I V, YAMASAKI M, DOEBLEY J F, MCMULLEN M D, GAUT B S, NIELSEN D M, HOLLAND J B, KRESOVICH S, BUCKLER E S. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nature Genetics, 2006, 38(2): 203-208.
[5] LIPPERT C, LISTGARTEN J, LIU Y, KADIE C M, DAVIDSON R I, HECKERMAN D. FaST linear mixed models for genome-wide association studies. Nature Methods, 2011, 8(10): 833-835.
[6] LOH P R, TUCKER G, BULIK-SULLIVAN B K, VILHJÁLMSSON B J, FINUCANE H K, SALEM R M, CHASMAN D I, RIDKER P M, NEALE B M, BERGER B, PATTERSON N, PRICE A L. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nature Genetics, 2015, 47(3): 284-290.
[7] JIANG L, ZHENG Z, QI T, KEMPER K E, WRAY N R, VISSCHER P M, YANG J. A resource-efficient tool for mixed model association analysis of large-scale data. Nature Genetics, 2019, 51(12): 1749-1755.
[8] 卜李那, 赵毅强. 全基因组关联分析及其扩展方法的研究进展. 农业生物技术学报, 2019, 27(1): 150-158.
BU L N, ZHAO Y Q. Research progress of genome-wide association study and its extension methods. Journal of Agricultural Biotechnology, 2019, 27(1): 150-158. (in Chinese)
[9] CARDON L R, PALMER L J. Population stratification and spurious allelic association. The Lancet, 2003, 361(9357): 598-604.
[10] DEVLIN B, ROEDER K. Genomic control for association studies. Biometrics, 1999, 55(4): 997-1004.
[11] PRICE A L, PATTERSON N J, PLENGE R M, WEINBLATT M E, SHADICK N A, REICH D. Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics, 2006, 38(8): 904-909.
[12] SHAM P C, PURCELL S M. Statistical power and significance testing in large-scale genetic studies. Nature Reviews Genetics, 2014, 15(5): 335-346.
[13] GAO X, BECKER L C, BECKER D M, STARMER J D, PROVINCE M A. Avoiding the high Bonferroni penalty in genome-wide association studies. Genetic Epidemiology, 2010, 34(1): 100-105.
[14] LIU X L, HUANG M, FAN B, BUCKLER E S, ZHANG Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. Plos Genetics, 2016, 12(2): 1-24.
[15] ZHAO K Y, ARANZANA M J, KIM S, LISTER C, SHINDO C, TANG C, TOOMAJIAN C, ZHENG H G, DEAN C, MARJORAM P, NORDBORG M. Anexample of association mapping in structured samples. Plos Genetics, 2007, 3(1): 71-82.
[16] XIAO Y J, LIU H J, WU L J, WARBURTON M, YAN J B. Genome-wide association studies in maize: Praise and stargaze. Molecular Plant, 2017, 10(3): 359-374.
[17] 温阳俊, 冯建英, 张瑾. 多位点关联分析方法学的研究进展. 南京农业大学学报, 2022, 45(1): 1-10.
WEN Y J, FENG J Y, ZHANG J. Research progress of multi-locus genome-wide association study. Journal of Nanjing Agricultural University, 2022, 45(1): 1-10. (in Chinese)
[18] KANG H M, ZAITLEN N A, WADE C M, KIRBY A, HECKERMAN D, DALY M J, ESKIN E. Efficient control of population structure in model organism association mapping. Genetics, 2008, 178(3): 1709-1723.
[19] KANG H M, SUL J H, SERVICE S K, ZAITLEN N A, KONG S Y, FREIMER N B, SABATTI C, ESKIN E. Variance component model to account for sample structure in genome-wide association studies. Nature Genetics, 2010, 42(4): 348-354.
[20] ZHANG Z W, ERSOZ E, LAI C Q, TODHUNTER R J, TIWARI H K, GORE M A, BRADBURY P J, YU J M, ARNETT D K, ORDOVAS J M, BUCKLER E S. Mixed linear model approach adapted for genome-wide association studies. Nature Genetics, 2010, 42(4): 355-360.
[21] LI M, LIU X L, BRADBURY P, YU J M, ZHANG Y M, TODHUNTER R J, BUCKLER E S, ZHANG Z W. Enrichment of statistical power for genome-wide association studies. BMC Biology, 2014, 12(1): 1-10.
[22] AULCHENKO Y S, de KONING D J, HALEY C. Genomewide rapid association using mixed model and regression: a fast and simple method for genomewide pedigree-based quantitative trait loci association analysis. Genetics, 2007, 177(1): 577-585.
[23] SVISHCHEVA G R, AXENOVICH T I, BELONOGOVA N M, van DUIJN C M, AULCHENKO Y S. Rapid variance components-based method for whole-genome association analysis. Nature Genetics, 2012, 44(10): 1166-1170.
[24] ZHOU X, STEPHENS M. Genome-wide efficient mixed-model analysis for association studies. Nature Genetics, 2012, 44(7): 821-824.
[25] LISTGARTEN J, LIPPERT C, KADIE C M, DAVIDSON R I, ESKIN E, HECKERMAN D. Improved linear mixed models for genome-wide association studies. Nature Methods, 2012, 9(6): 525-526.
[26] WANG Q S, TIAN F, PAN Y C, BUCKLER E S, ZHANG Z W. A SUPER powerful method for genome wide association study. Plos One, 2014, 9(9): 1-9.
[27] BULIK-SULLIVAN B K, LOH P R, FINUCANE H K, RIPKE S, YANG J, PATTERSON N, DALY M J, PRICE A L, NEALE B M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nature Genetics, 2015, 47(3): 291-295.
[28] SEGURA V, VILHJÁLMSSON B J, PLATT A, KORTE A, SEREN Ü, LONG Q, NORDBORG M. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nature Genetics, 2012, 44(7): 825-830.
[29] HUANG M, LIU X L, ZHOU Y, SUMMERS R M, ZHANG Z W. BLINK: a package for the next level of genome-wide association studies with both individuals and markers in the millions. GigaScience, 2019, 8(2): 1-12.
[30] FU W W, WANG R, XU N Y, WANG J X, LI R, NANAEI H A, NIE Q H, ZHAO X, HAN J L, YANG N, JIANG Y. Galbase: A comprehensive repository for integrating chicken multi-omics data. BMC Genomics, 2022, 23(1): 1-11.
[31] YANG J, LEE S H, GODDARD M E, VISSCHER P M. GCTA: A tool for genome-wide complex trait analysis. American Journal of Human Genetics. 2011, 88(1): 76-82.
[32] CHEN H, WANG C L, CONOMOS M P, STILP A M, LI Z L, SOFER T, SZPIRO A A, CHEN W, BREHM J M, CELEDON J C, REDLINE S, PAPANICOLAOU G J, THORNTON T A, LAURIE C C, RICE K, LIN X H. Control for population structure and relatedness for binary traits in genetic association studies via logistic mixed models. The American Journal of Human Genetics, 2016, 98(4): 653-666.
[33] ZHOU W, NIELSEN J B, FRITSCHE L G, DEY R, GABRIELSEN M E, WOLFORD B N, LEFAIVE J, VANDEHAAR P, GAGLIANO S A, GIFFORD A, BASTARACHE L A, WEI W Q, DENNY J C, LIN M X, HVEEM K, KANG H M, ABECASIS G R, WILLER C J, LEE S. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nature Genetics, 2018, 50(9): 1335-1341.
[34] JIANG L D, ZHENG Z L, FANG H L, YANG J. A generalized linear mixed model association tool for biobank-scale data. Nature Genetics, 2021, 53(11): 1616-1621.
[35] BI W J, ZHOU W, DEY R, MUKHERJEE B, SAMPSON J N, LEE S. Efficient mixed model approach for large-scale genome-wide association studies of ordinal categorical phenotypes. The American Journal of Human Genetics, 2021, 108(5): 825-839.
[36] HANSEN M, KRAFT T, GANESTAM S, SÄLL T, NILSSON N O. Linkage disequilibrium mapping of the bolting gene in sea beet using AFLP markers. Genetical Research, 2001, 77(1): 61-66.
[37] ATWELL S, HUANG Y S, VILHJÁLMSSON B J, WILLEMS G, HORTON M, LI Y, MENG D Z, PLATT A, TARONE A M, HU T T, JIANG R, MULIYATI N W, ZHANG X, AMER M A, BAXTER I, BRACHI B, CHORY J, DEAN C, DEBIEU M, de MEAUX J, ECKER J R, FAURE N, KNISKERN J M, JONES J D G, MICHAEL T, NEMRI A, ROUX F, SALT D E, TANG C L, TODESCO M, TRAW M B, WEIGEL D, MARJORAM P, BOREVITZ J O, BERGELSON J, NORDBORG M. Genome-wide association study of 107 phenotypes ininbred lines. Nature, 2010, 465(7298): 627-631.
[38] ZHAO K Y, TUNG C W, EIZENGA G C, WRIGHT M H, ALI M L, H PRICE A, NORTON G J, ISLAM M R, REYNOLDS A, MEZEY J, MCCLUNG A M, BUSTAMANTE C D, MCCOUCH S R. Genome-wide association mapping reveals a rich genetic architecture of complex traits in. Nature Communications, 2011, 2(1): 1-10.
[39] HUANG X H, WEI X H, SANG T, ZHAO Q, FENG Q, ZHAO Y, LI C Y, ZHU C R, LU T T, ZHANG Z W, LI M, FAN D L, GUO Y L, WANG A H, WANG L, DENG L W, LI W J, LU Y Q, WENG Q J, LIU K Y, HUANG T, ZHOU T Y, JING Y F, LI W, LIN Z, BUCKLER E S, QIAN Q, ZHANG Q F, LI J Y, HAN B. Genome-wide association studies of 14 agronomic traits in rice landraces. Nature Genetics, 2010, 42(11): 961-967.
[40] HUANG X H, ZHAO Y, WEI X H, LI C Y, WANG A H, ZHAO Q, LI W J, GUO Y L, DENG L W, ZHU C R, FAN D L, LU Y Q, WENG Q J, LIU K Y, ZHOU T F, JING Y F, SI L Z, DONG G J, HUANG T, LU T T, FENG Q, QIAN Q, LI J Y, HAN B. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nature Genetics, 2012, 44(1): 32-39.
[41] YONEMARU J I, MIZOBUCHI R, KATO H, YAMAMOTO T, YAMAMOTO E, MATSUBARA K, HIRABAYASHI H, TAKEUCHI Y, TSUNEMATSU H, ISHII T, OHTA H, MAEDA H, EBANA K, YANO M. Genomic regions involved in yield potential detected by genome-wide association analysis in Japanese high-yielding rice cultivars. BMC genomics, 2014, 15(1): 1-12.
[42] WANG M, YAN J B, ZHAO J R, SONG W, ZHANG X B, XIAO Y N, ZHENG Y L. Genome-wide association study (GWAS) of resistance to head smut in maize. Plant Science, 2012, 196(1): 125-131.
[43] LI H, PENG Z Y, YANG X H, WANG W D, FU J J, WANG J H, HAN Y J, CHAI Y C, GUO T T, YANG N, LIU J, WARBURTON M L, CHENG Y B, HAO X M, ZHANG P, ZHAO J Y, LIU Y J, WANG G Y, LI J S, YAN J B. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nature Genetics, 2013, 45(1): 43-50.
[44] LI Y X, LI C H, BRADBURY P J, LIU X L, LU F, ROMAY C M, GLAUBITZ J C, WU X, PENG B, SHI Y S, SONG Y C, ZHANG D F, BUCKLER E S, ZHANG Z W, LI Y, WANG T Y. Identification of genetic variants associated with maize flowering time using an extremely large multi-genetic background population. The Plant Journal, 2016, 86(5): 391-402.
[45] SEKHON R S, SASKI C, KUMAR R, FLINN B S, LUO F, BEISSINGER T M, ACKERMAN A J, BREITZMAN M W, BRIDGES W C, DE LEON N, KAEPPLER S M. Integrated genome-scale analysis identifies novel genes and networks underlying senescence in maize. The Plant Cell, 2019, 31(9): 1968-1989.
[46] CHAO Z F, CHEN Y Y, JI C, WANG Y L, HUANG X, ZHANG C Y, YANG J, SONG T, WU J C, GUO L X, LIU C B, HAN M L, WU Y R, YAN J B, CHAO D Y. A genome-wide association study identifies a transporter for zinc uploading to maize kernels. Embo Reports, 2023, 24(1): 1-19.
[47] REN J, DUAN Y Y, QIAO R M, YAO F, ZHANG Z Y, YANG B, GUO Y M, XIAO S J, WEI R X, OUYANG Z X, DING N S, AI H S, HUANG L S. A missense mutation in PPARD causes a major QTL effect on ear size in pigs. Plos Genetics, 2011, 7(5): 1-10.
[48] MA J W, YANG J, ZHOU L S, REN J, LIU X X, ZHANG H, YANG B, ZHANG Z Y, MA H B, XIE X H, XING Y Y, GUO Y M, HUANG L S. A splice mutation in the PHKG1 gene causes high glycogen content and low meat quality in pig skeletal muscle. Plos Genetics, 2014, 10(10): 1-13.
[49] WANG X M, LIU X L, DENG D D, YU M, LI X P. Genetic determinants of pig birth weight variability. BMC Genetics, 2016, 17(1): 41-48.
[50] GUO X Y, SU G S, CHRISTENSEN O F, JANSS L, LUND M S. Genome-wide association analyses using a Bayesian approach for litter size and piglet mortality in Danish Landrace and Yorkshire pigs. BMC Genomics, 2016, 17(1): 1-12.
[51] GOZALO-MARCILLA M, BUNTJER J, JOHNSSON M, BATISTA L, DIEZ F, WERNER C R, CHEN C Y, GORJANC G, MELLANBY R J, HICKEY J M, ROS-FREIXEDES R. Genetic architecture and major genes for backfat thickness in pig lines of diverse genetic backgrounds. Genetics, Selection, Evolution, 2021, 53(1): 1-14.
[52] GU X R, FENG C G, MA L, SONG C, WANG Y Q, DA Y, LI H F, CHEN K W, YE S H, GE C R, HU X X, LI N. Genome-wide association study of body weight in chicken F2resource population. Plos One, 2011, 6(7): 1-5.
[53] IMSLAND F, FENG C G, BOIJE H, BED'HOM B, FILLON V, DORSHORST B, RUBIN C J, LIU R R, GAO Y, GU X R, WANG Y Q, GOURICHON D, ZODY M C, ZECCHIN W, VIEAUD A, TIXIER-BOICHARD M, HU X X, HALLBÖÖK F, LI N, ANDERSSON L. The Rose-comb mutation in chickens constitutes a structural rearrangement causing both altered comb morphology and defective sperm motility. Plos Genetics, 2012, 8(6): 1-12.
[54] GUO Y, GU X R, SHENG Z Y, WANG Y Q, LUO C L, LIU R R, QU H, SHU D M, WEN J, CROOIJMANS R P M A, CARLBORG Ö, ZHAO Y Q, HU X X, LI N. A complex structural variation on chromosome 27 leads to the ectopic expression of hoxb8 and the muffs and beard phenotype in chickens. Plos Genetics, 2016, 12(6): 1-24.
[55] WANG Y Z, CAO X M, LUO C L, SHENG Z Y, ZHANG C Y, BIAN C, FENG C G, LI J X, GAO F, ZHAO Y Q, JIANG Z Q, QU H, SHU D M, CARLBORG Ö, HU X X, LI N. Multiple ancestral haplotypes harboring regulatory mutations cumulatively contribute to a QTL affecting chicken growth traits. Communications Biology, 2020, 3(1): 1-13.
[56] FAN Q C, WU P F, DAI G J, ZHANG G X, ZHANG T, XUE Q, SHI H Q, WANG J Y. Identification of 19 loci for reproductive traits in a local Chinese chicken by genome-wide study. Genetics and Molecular Research, 2017, 16(1): 1-8.
[57] LI Q L, DUAN Z Y, SUN C J, ZHENG J X, XU G Y, YANG N. Genetic variations for the eggshell crystal structure revealed by genome-wide association study in chickens. BMC Genomics, 2021, 22(1): 1-12.
[58] GUO Y P, HUANG H T, ZHANG Z Z, MA Y C, LI J Z, TANG H H, MA H X, LI Z J, LI W T, LIU X J, KANG X T, HAN R L. Genome-wide association study identifies SNPs for growth performance and serum indicators inbroilers () using ddGBS sequencing. BMC Genomics, 2022, 23(1): 1-11.
[59] 张统雨, 朱才业, 杜立新, 赵福平. 羊重要性状全基因组关联分析研究进展. 遗传, 2017, 39(06): 491-500.
ZHANG T Y, ZHU C Y, DU L X, ZHAO F P. Advances in genome-wide association studies for important traits in sheep and goats. Hereditas(Beijing), 2017, 39(6): 491-500. (in Chinese)
[60] DEMARS J, FABRE S, SARRY J, ROSSETTI R, GILBERT H, PERSANI L, TOSSER-KLOPP G, MULSANT P, NOWAK Z, DROBIK W, MARTYNIUK E, BODIN L. Genome-wide association studies identify two novel BMP15 mutations responsible for an atypical hyperprolificacy phenotype in sheep. Plos Genetics, 2013, 9(4): 1-13.
[61] HE X H, ZHOU Z K, PU Y B, CHEN X F, MA Y H, JIANG L. Mapping the four-horned locus and testing the polled locus in three Chinese sheep breeds. Animal Genetics, 2016, 47(5): 623-627.
[62] GJEDREM T. Genetic improvement for the development of efficient global aquaculture: A personal opinion review. Aquaculture, 2012, 344-349(1): 12-22.
[63] HOLBORN M K, ANG K P, ELLIOTT J A K, POWELL F, BOULDING E G. Genome wide association analysis for bacterial kidney disease resistance in a commercial North American Atlantic salmon () population using a 50K SNP panel. Aquaculture, 2018, 495(1): 465-471.
[64] PENG W Z, XU J, ZHANG Y, FENG J X, DONG C J, JIANG L K, FENG J Y, CHEN B H, GONG Y W, CHEN L, XU P. An ultra-high density linkage map and QTL mapping for sex and growth-related traits of common carp (). Scientific Reports, 2016, 6(1): 1-16.
[65] LIN H L, ZHOU Z X, ZHAO J, ZHOU T, BAI H Q, KE Q Z, PU F, ZHENG W Q, XU P. Genome-wide association study identifies genomic loci of sex determination and gonadosomatic index traits in large yellow croaker (). Marine Biotechnology, 2021, 23(1): 127-139.
[66] DAVILA OLIVAS N H, KRUIJER W, GORT G, WIJNEN C L, VAN LOON J J A, DICKE M. Genome-wide association analysis reveals distinct genetic architectures for single and combined stress responses in. New Phytologist, 2017, 213(2): 838-851.
[67] ZHANG F, HU Z Q, WU Z C, LU J L, SHI Y Y, XU J L, WANG X Y, WANG J P, ZHANG F, WANG M M, SHI X R, CUI Y R, VERA CRUZ C, ZHUO D L, HU D D, LI M, WANG W S, ZHAO X Q, ZHENG T Q, FU B Y, ALI J, ZHOU Y L, LI Z K. Reciprocal adaptation of rice andpv.: cross-species 2D GWAS reveals the underlying genetics. The Plant Cell, 2021, 33(8): 2538-2561.
[68] MARTINS L B, BALINT-KURTI P, REBERG-HORTON S C. Genome-wide association study for morphological traits and resistance to Peryonella pinodes in the USDA pea single plant plus collection. G3 Genes|Genomes|Genetics, 2022, 12(9): 1-8.
[69] KARASOV T L, CHAE E, HERMAN J J, BERGELSON J. Mechanisms to mitigate the trade-off between growth and defense. The Plant Cell, 2017, 29(4): 666-680.
[70] DE RONNE M, SANTHANAM P, CINGET B, LABBÉ C, LEBRETON A, YE H, VUONG T D, HU H F, VALLIYODAN B, EDWARDS D, NGUYEN H T, BELZILE F, BÉLANGER R. Mapping of partial resistance to Phytophthora sojae in soybean PIs using whole-genome sequencing reveals a major QTL. The Plant Genome, 2022, 15(1): 1-16.
[71] LIU Q, CHENG L, NIAN H, JIN J, LIAN T X. Linking plant functional genes to rhizosphere microbes: A review. Plant Biotechnology Journal, 2023, 21(5): 902-917.
[72] BAI B, LIU W D, QIU X Y, ZHANG J, ZHANG J Y, BAI Y. The root microbiome: Community assembly and its contributions to plant fitness. Journal of Integrative Plant Biology, 2022, 64(2): 230-243.
[73] BERGELSON J, MITTELSTRASS J, HORTON M W. Characterizing both bacteria and fungi improves understanding of theroot microbiome. Scientific Reports, 2019, 9(1): 1-11.
[74] DENG S W, CADDELL D F, XU G, DAHLEN L, WASHINGTON L, YANG J L, COLEMAN-DERR D. Genome wide association study reveals plant loci controlling heritability of the rhizosphere microbiome. The ISME Journal, 2021, 15(11): 3181-3194.
[75] de FREITAS A S, de DAVID D B, TAKAGAKI B M, ROESCH L F W. Microbial patterns in rumen are associated with gain of weight in beef cattle. Antonie Van Leeuwenhoek, 2020, 113(9): 1299-1312.
[76] MALTECCA C, BERGAMASCHI M, TIEZZI F. The interaction between microbiome and pig efficiency: A review. Journal of Animal Breeding and Genetics, 2020, 137(1): 4-13.
[77] XUE M Y, SUN H Z, WU X H, LIU J X, GUAN L L. Multi-omics reveals that the rumen microbiome and its metabolome together with the host metabolome contribute to individualized dairy cow performance. Microbiome, 2020, 8(1): 1-19.
[78] ELOKIL A A, MAGDY M, MELAK S, ISHFAQ H, BHUIYAN A, CUI L, JAMIL M, ZHAO S, LI S. Faecal microbiome sequences in relation to the egg-laying performance of hens using amplicon-based metagenomic association analysis. Animal, 2020, 14(4): 706-715.
[79] CRESPO-PIAZUELO D, MIGURA-GARCIA L, ESTELLÉ J, CRIADO-MESAS L, REVILLA M, CASTELLÓ A, MUÑOZ M, GARCÍA-CASCO J M, FERNÁNDEZ A I, BALLESTER M, FOLCH J M. Association between the pig genome and its gut microbiota composition. Scientific Reports, 2019, 9(1): 1-11.
[80] BERGAMASCHI M, MALTECCA C, SCHILLEBEECKX C, MCNULTY N P, SCHWAB C, SHULL C, FIX J, TIEZZI F. Heritability and genome-wide association of swine gut microbiome features with growth and fatness parameters. Scientific Reports, 2020, 10(1): 1-12.
[81] WANG Y T, SUNG P Y, LIN P L, YU Y W, CHUNG R H. A multi-SNP association test for complex diseases incorporating an optimal P-value threshold algorithm in nuclear families. BMC Genomics, 2015, 16(1): 1-10.
[82] WANG F, MEYER N J, WALLEY K R, RUSSELL J A, FENG R. Causal genetic inference using haplotypes as instrumental variables. Genetic Epidemiology, 2016, 40(1): 35-44.
[83] N'DIAYE A, HAILE J K, CORY A T, CLARKE F R, CLARKE J M, KNOX R E, POZNIAK C J. Single marker and haplotype-based association analysis of semolina and pasta colour in elite durum wheat breeding lines using a high-density consensus map. Plos One, 2017, 12(1): 1-24.
[84] YANO K, YAMAMOTO E, AYA K, TAKEUCHI H, LO P C, HU L, YAMASAKI M, YOSHIDA S, KITANO H, HIRANO K, MATSUOKA M. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nature Genetics, 2016, 48(8): 927-934.
[85] OGAWA D, YAMAMOTO E, OHTANI T, KANNO N, TSUNEMATSU H, NONOUE Y, YANO M, YAMAMOTO T, YONEMARU J I. Haplotype-based allele mining in the Japan-MAGIC rice population. Scientific Reports, 2018, 8(1): 1-11.
[86] ZHANG Z, GUILLAUME F, SARTELET A, CHARLIER C, GEORGES M, FARNIR F, DRUET T. Ancestral haplotype-based association mapping with generalized linear mixed models accounting for stratification. Bioinformatics, 2012, 28(19): 2467-2473.
[87] ZHANG H, SHEN L Y, XU Z C, KRAMER L M, YU J Q, ZHANG X Y, NA W, YANG L L, CAO Z P, LUAN P, REECY J M, LI H. Haplotype-based genome-wide association studies for carcass and growth traits in chicken. Poultry Science, 2020, 99(5): 2349-2361.
[88] HOWARD D M, HALL L S, HAFFERTY J D, ZENG Y N, ADAMS M J, CLARKE T K, PORTEOUS D J, NAGY R, HAYWARD C, SMITH B H, MURRAY A D, RYAN N M, EVANS K L, HALEY C S, DEARY I J, THOMSON P A, MCINTOSH A M. Genome-wide haplotype-based association analysis of major depressive disorder in Generation Scotland and UK Biobank. Translational Psychiatry, 2017, 7(11): 1-9.
Principle, Optimization and Application of Mixed Models in Genome- Wide Association Study
College of Biological Sciences, China Agricultural University, Beijing 100193
Genome-wide association study (GWAS) is an effective method to locate genomic loci that are significantly associated with traits. With the accumulated phenotypic data, the continuous development of high-throughput genotyping technology, and the improved statistical methods, it promotes the wide application of GWAS in area of human disease and animal and plant genetics. False positives are one of the important concerns that impair the reliability of genome-wide association results. To control the false positives, in addition to correcting the-values, GWAS models have been continuously improved from the naive methods like ANOVA (for quantitative trait) or Chi-square test (for quality trait), to general linear model (GLM), which incorporates fixed-effect covariates, to the mixed linear model (MLM), which incorporates random effects. Fitting individual genetic effects into random effects defined by the genomic relationships matrix (GRM) is commonly adapted currently. Since the parameter estimation of MLM consumes a lot of computational resources, researchers have tried to optimize solving models and constructing GRM (which also improves computing efficiency), and the time complexity gradually decreased from O(MN3) to O(MN) for MLM-based methods, achieving a great leap in computational speed and statistical efficacy. For inflations caused by unbalanced case-control data, researchers further correct the generalized mixed linear model (GLMM). This paper comprehensively introduces the basic principles and development of GWAS, with specific emphasis on the model improvement and optimization details. We also list the applications of MLM in GWAS in agriculture, including progress on animals, plants and microbes, as well as the application of haplotype in GWAS. Finally, we give prospects on the future developments of GWAS from the viewpoints of further model optimization and experimental design.
genome-wide association study; complex traits; random effects; genomic relationships matrix; mixed linear model
2022-12-04;
2023-03-02
国家重点研发计划(2022YFF1000204)
谭力治,E-mail:tanlizhi@cau.edu.cn。通信作者赵毅强,E-mail:yiqiangz@cau.edu.cn
10.3864/j.issn.0578-1752.2023.09.001
(责任编辑 李莉)
