下调MAC30对乳腺癌细胞焦亡、血管生成及ATP1A2蛋白的作用机制
2023-06-09潘蕾蕾马俊叶亭亭陈剑平宣城中心医院心胸乳腺外科安徽宣城4000皖南医学院弋矶山医院甲乳外科安徽芜湖4000
潘蕾蕾,马俊,叶亭亭,陈剑平 (. 宣城中心医院心胸乳腺外科,安徽 宣城 4000;. 皖南医学院弋矶山医院甲乳外科,安徽 芜湖 4000)
乳腺癌是女性最常见的恶性肿瘤之一,2020年乳腺癌新增人数约226万,已经超过肺癌成为全球发病率最高的恶性肿瘤[1]。手术及放化疗是临床治疗乳腺癌的主要方法,但放疗抵抗或化疗耐药会影响患者疗效及预后[2]。细胞焦亡又称细胞炎性坏死,主要依赖Gasdermin(GSDM)蛋白调节细胞生物活性。有研究表示,caspase-3可通过切割GSDM蛋白N端结构增加细胞焦亡,从而降低癌细胞化疗耐药性[3]。乳腺癌细胞在缺血环境下无法进行增殖及转移,肿瘤血管生成可为癌细胞活化提供血供,从而促进乳腺癌疾病进展。因此,众多学者认为可将抑制肿瘤血管生成作为抗乳腺癌的靶点[4-5]。现阶段,在血管新生的病理生理过程中,细胞自噬作用持续存在,在血管内皮生长因子(vascular endothelial growth factor,VEGF)诱导的血管生成中,自噬相关信号可通过调控血管内皮细胞表面内皮生长因子受体2(vascular endothelial growth factor receptor 2,VEGFR2)表达而促进血管生成。有研究发现,Na+-K+-ATP酶的α亚单位在肿瘤中存在异常表达,对癌细胞增殖及迁移等具有调控作用[6]。Na+-K+-ATP酶α2亚基(ATP1A2)基因在乳腺癌中高表达,其功能缺失会使细胞内Ca2+增多,从而参与癌症的发生发展。但ATP1A2基因与肿瘤血管生成及细胞自噬的关系目前尚不清楚。
脑膜瘤相关蛋白(meningioma-associated protein,MAC30)是胰岛素生长因子结合蛋白家族成员之一,主要存在于细胞核的内质网中,并在机体组织中广泛表达[7]。在乳腺癌组织中MAC30表达升高,通过下调MAC30能够抑制肿瘤细胞侵袭及迁移,但是关于下调MAC30对乳腺癌细胞焦亡、血管生成及ATP1A2蛋白水平的影响目前尚未可知。因此,本研究通过体外培养乳腺癌细胞,观察下调MAC30对乳腺癌的改善作用,以期为其治疗提供参考。
1 材料与方法
1.1 细胞来源及细胞培养
乳腺上皮细胞HMEC和乳腺癌细胞MDA-MB231、MCF-7、SKBR3均购自中国科学院典型培养物保藏中心,于37 ℃、5%CO2饱和湿度下培养,24 h后弃去培养基,置于含1%双抗的10%胎牛血清中培养,培养条件同上,细胞生长至80%~90%时进行传代,生长至90%时终止消化反应,培养基中2~3 d传代。
1.2 试剂与仪器来源
MAC30 Lenti-shRNA和阴性对照Lenti-shRNA(NC-shRNA)由上海吉凯基因公司合成;RPMI 1640培养基购自武汉益普生物科技有限公司;ATP1A2多克隆抗体购自武汉艾美捷科技有限公司;鼠抗人GAPDH单克隆抗体购自上海圻明生物科技有限公司;山羊抗小鼠IgG购自武汉纯度生物科技有限公司;CCK-8法试剂盒购自上海Beyotime公司;AnnexinV-FITC/PI凋亡检测试剂盒购自上海康朗生物科技有限公司;ECL显色试剂盒购自北京索宝莱科技有限公司;BCA蛋白定量试剂盒购自上海雅酶生物医药科技有限公司;酶标仪(型号:HBS-1096A)购自上海研卉生物科技有限公司;TDZ4-WS低速台式离心机购自湖南湘瑞生物有限公司;凝胶成像系统及Western blot电泳系统均购自北京赛百奥科技有限公司;荧光定量PCR仪购自中山大学达安基因股份有限公司。
1.3 qRT-PCR检测HMEC、MDA-MB231、MCF-7和SKBR3细胞MAC30 mRNA表达
使用TRIzol法提取HMEC、MDA-MB231、MCF-7和SKBR3细胞中总RNA,无核酸酶溶解后转录为cDNA,获得反应体系,条件为:42 ℃ 60 min,72 ℃ 5 min,4 ℃终点。采用qRT-PCR检测,每个细胞设置6个复孔,上下游引物均为0.5 μL,反应条件:95 ℃预热3 min,95 ℃ 5 s,58 ℃退火,40个循环。引物序列:MAC30上游为5'-CTCAGCCACATCCCC-3',下游为5'-CACAAGCTCG CAAAAC-3';β-actin上游为5'-GGAAATCGTGCGTGACATTA-3',下游为5'-GGAGCAATGATCTTG ATCTTC-3'。根据2-△△Ct法计算各组细胞中MAC30相对表达量。
1.4 MCF-7细胞分组
取MCF-7细胞,待生长至50%~60%后采用培养基制成单个细胞悬液,分为乳腺癌组(MCF-7细胞)、NC组(MCF-7细胞转染MAC30-NC-shRNA)、MAC30 shRNA组(MCF-7细胞转染MAC30-shRNA)、ATP1A2组(MCF-7细胞转染ATP1A2-shRNA)及MAC30 shRNA+ATP1A2组(MCF-7细胞转染MAC30-shRNA、ATP1A2-shRNA)。
1.5 MAC30 shRNA慢病毒转染
将生长状态良好的MCF-7细胞制备成5×107/mL的细胞悬液,接种至6孔板中,37 ℃孵育24 h,待细胞达到30%时更换培养基,加入感染增强液,根据最佳感染复数及病毒滴度,加入MAC30 Lenti-shRNA和阴性对照NC-shRNA培养液,次日换掉病毒培养液,加入1 mg新鲜培养基,96 h后采用qRT-PCR验证转染效率。
1.6 ATP1A2质粒转染
构建TP1A2质粒[7],将MCF-7细胞接种于6孔板中,待细胞生长覆盖孔板80%~90%时开始转染,配制转染混合液,将4 μg质粒加入500 μL无血清培养基中,将5 μL ip 2000加入500 μL无血清培养液混匀,室温下静置20 min。PBS冲洗3次,加入1 mL Lipo⁃fectamine 2000转染混合液及无血清培养基1.5 mL,培养6 h后,换成常规培养基继续培养48 h。
1.7 MCF-7细胞增殖实验
采用CCK-8法检测各组MCF-7细胞增殖情况。将5×103个对数生长的MCF-7细胞接种至96孔板中,贴壁培养24 h后,加入CCK-8试剂(5 g/L),每孔20 μL,再加入200 μL二甲基亚砜溶液,充分溶解15 min,分别于培养24 h、48 h、72 h、96 h时取出培养板,在酶标仪450 nm波长处检测吸光度OD值,实验3次,复孔2次。
1.8 MCF-7细胞焦亡实验
100 μL的Binding buffer重悬细胞后加入5 μL AnnexinV-FITC和5 μL PI混合液,室温避光孵育15 min,再加入400 μL含Binding buffer的细胞悬液,30 min后使用流式细胞仪进行检测。以AnnexinV-FITC+/PI+双阳性细胞百分率作为焦亡率。实验重复3次,取平均值。
1.9 体外血管形成实验
取48孔板,于冰上每孔加入60 μL基质胶溶液,然后在37 ℃、5%CO2的培养箱静置30 min使其凝固,取各组细胞进行消化、离心,弃掉上清液后重悬细胞,调整密度为2×105/mL接种于24孔板,在37 ℃、5%CO2的培养箱中进行培养,48 h后置于倒置显微镜下观察。计算5个视野的血管管腔数目,实验重复3次,取平均值。
1.10 Westerm blot检测各组细胞ATP1A2、SCr、AKT及PI3K水平
10%SDS-聚丙烯酰胺凝胶分离蛋白(40 μg),转移至PVDF膜上后再用5%脱脂奶粉封闭2~3 h,加入一抗ATP1A2(1∶100)、SCr(1∶200)、AKT(1∶150)、PI3K(1∶200),内参为GAPDH(1∶2 000),4 ℃摇床孵育过夜。TBST冲洗3次,室温下与辣根过氧化物酶标记山羊抗小鼠IgG(1∶5 000)结合孵育2 h,杂交冲洗。然后将膜浸入ECL工作液,计算目标蛋白与GAPHD之间的OD值,检测相对表达。实验重复3次,取平均值。
1.11 统计学分析
采用GraphPad Prism 8软件分析数据,各组数据服从正态分布,计量资料以均数±标准差()表示,多组间比较采用方差分析,事后检验采用Bonferroni法进行组间两两比较,P<0.05表示差异有统计学意义。
2 结果
2.1 HMEC、MDA-MB231、MCF-7和SKBR3细胞MAC30 mRNA表达
与HMEC细胞相比,MDA-MB231、MCF-7和SKBR3细胞MAC30 mRNA表达水平均升高(P<0.05),其中MCF-7细胞MAC30 mRNA表达水平最高,见图1。故本研究选择MCF-7细胞进行后续实验。

图1 HMEC、MDA-MB231、MCF-7和SKBR3细胞MAC30 mRNA表达
2.2 各组MCF-7细胞中MAC30 mRNA表达
乳腺癌组与NC组MAC30 mRNA表达水平比较,差异无统计学意义(P>0.05);与乳腺癌组和NC组相比,MAC30 shRNA组MAC30 mRNA表达水平降低(P<0.05);与MAC30 shRNA组相比,ATP1A2组MAC30 mRNA表达水平升高(P<0.05);与ATP1A2组相比,MAC30 shRNA+ATP1A2组MAC30 mRNA表达水平降低(P<0.05),见图2。
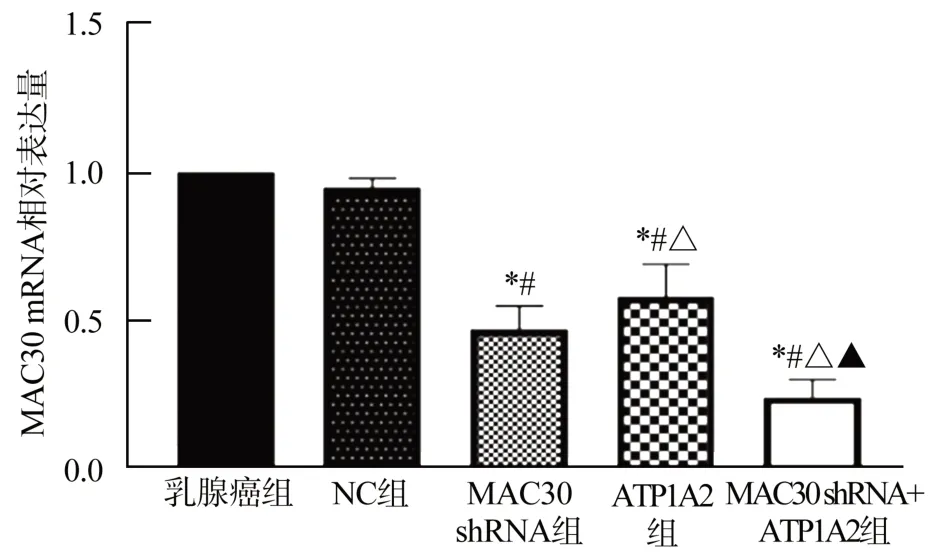
图2 各组MCF-7细胞中MAC30 mRNA表达
2.3 各组MCF-7细胞增殖活性比较
24 h、48 h、72 h及96 h时,乳腺癌组与NC组MCF-7细胞增殖活性比较差异均无统计学意义(P>0.05);与乳腺癌组和NC组相比,MAC30 shRNA组、ATP1A2组及MAC30 shRNA+ATP1A2组细胞增殖活性均降低(P<0.05);MAC30 shRNA组与ATP1A2组细胞增殖活性比较差异无统计学意义(P>0.05);与MAC30 shRNA组和ATP1A2组相比,MAC30 shRNA+ATP1A2组细胞增殖活性降低(P<0.05),见图3。

图3 各组MCF-7细胞增殖活性
2.4 各组MCF-7细胞焦亡率比较
MAC30 shRNA及ATP1A2质粒均可以诱发MCF-7细胞形态改变,主要体现在细胞体积缩小、细胞颗粒增多及大量焦亡。细胞之间连接较少,以MAC30 shRNA+ATP1A2组最佳,培养液出现大量细胞悬浮,见图4a。乳腺癌组与NC组MCF-7细胞焦亡率比较,差异无统计学意义(P>0.05);与乳腺癌组和NC组比较,MAC30 shRNA组、ATP1A2组及MAC30 shRNA+ATP1A2组MCF-7细胞焦亡率均升高,其中MAC30 shRNA+ATP1A2组最高,而MAC30 shRNA组与ATP1A2组比较,差异无统计学意义(P>0.05),见图4b。
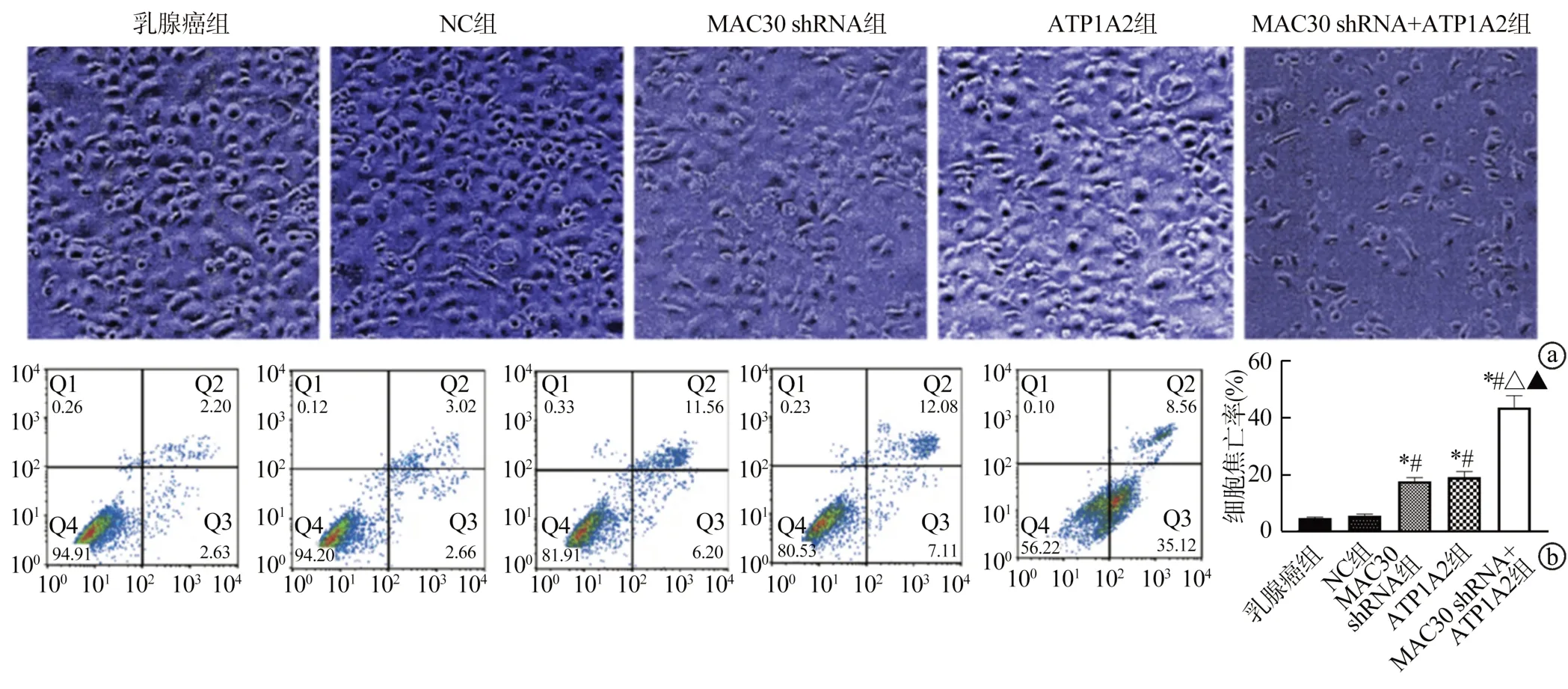
图4 各组MCF-7细胞形态及细胞焦亡率比较
2.5 各组MCF-7细胞血管形成数目比较
乳腺癌组与NC组细胞血管形成数目比较差异无统计学意义(P>0.05);与乳腺癌组和NC组相比,MAC30 shRNA组、ATP1A2组及MAC30 shRNA+ATP1A2组细胞血管形成数目减少(P<0.05),MAC30 shRNA组与ATP1A2组细胞血管形成数目比较差异无统计学意义(P>0.05);与MAC30 shRNA组和ATP1A2组相比,MAC30 shRNA+ATP1A2组细胞血管形成数目减少(P<0.05),见图5。

图5 各组MCF-7细胞血管形成比较( ×200)
2.6 各组MCF-7细胞ATP1A2、SCr、AKT及PI3K水平比较
乳腺癌组与NC组细胞ATP1A2、SCr、AKT及PI3K表达水平比较差异无统计学意义(P>0.05);与乳腺癌组和NC组相比,MAC30 shRNA组、ATP1A2组及MAC30 shRNA+ATP1A2组ATP1A2、SCr、AKT及PI3K表达水平降低(P<0.05);MAC30 shRNA组与ATP1A2组ATP1A2、SCr、AKT及PI3K表达水平比较差异无统计学意义(P>0.05);与MAC30 shRNA组和ATP1A2组相比,MAC30 shRNA+ATP1A2组ATP1A2、SCr、AKT及PI3K表达水平降低(P<0.05),见图6。

图6 各组MCF-7细胞ATP1A2、SCr、AKT及PI3K水平比较
3 讨论
乳腺癌是我国发病率居首位的恶性肿瘤[8]。研究其治疗机制对于提高临床治疗效果、延长患者预后具有重要作用。本研究通过观察下调MAC30对乳腺癌细胞焦亡、血管生成及ATP1A2蛋白水平的影响,为乳腺癌的治疗提供参考。
MAC30是胰岛素样生长因子结合蛋白家族成员之一,位于染色体17q11.2,具有调节胰岛素样生长因子(insulin-like growth factor,IGF)活性的作用[9]。MAC30在人体内(包括肝细胞、支气管上皮细胞、巨噬细胞和树突状细胞等)广泛分布,在某些癌细胞和癌组织中也有表达[10]。MAC30在不同恶性肿瘤中的表达存在差异,如在胰腺癌[11]中呈低表达,而在胃癌[11]、结肠癌[11]及卵巢癌[12]中呈高表达。MAC30可能参与乳腺癌的生长和转移。乳腺癌组织中MAC30的表达高于正常组织。此外,肿瘤细胞表面的MAC30与癌细胞的浸润能力和转移能力有关。因此,MAC30也被认为是开发抗肿瘤药物的一个潜在靶点。在乳腺癌中MAC30过表达可被乳腺癌易感基因1(breast cancer1,BRCA1)诱导后突变,进而加快疾病进展。MAC30作为细胞内孤儿受体,可与sigma2受体结合进而促进癌细胞生长及分化[13]。本研究结果表明,在乳腺癌细胞中下调MAC30可抑制癌细胞增殖,加快焦亡。有研究证实,在胃癌细胞中抑制MAC30可降低蛋白激酶B(protein kinase B,PKB)/AKT磷酸化及细胞周期蛋白B1(cyclin B1)活化,进而降低胃癌细胞活性,推测MAC30表达与肿瘤细胞活性相关[14]。细胞焦亡作为细胞程序性死亡的方式之一,当细胞焦亡发生时GSDMD活性升高,可通过促进细胞膜裂解而加快肿瘤细胞死亡[15]。下调MAC30可加快癌细胞焦亡,推测机制为抑制MAC30后磷脂酰肌醇3激酶(phosphatidylinositol 3-kinase,PI3K)/AKT受到抑制,可促进乳腺癌细胞焦亡,发挥抗肿瘤作用。
当肿瘤体积较小时,由于肿瘤细胞对营养物质和氧气等的需求量相对较低,可以通过弥散方式从周围组织中摄取所需的营养物质。但当肿瘤体积增大到一定程度时,弥散方式无法满足肿瘤细胞的需求,肿瘤细胞就会陷入缺氧状态。为了维持生长,肿瘤细胞开始释放一系列生长因子(如VEGF等)来刺激周围血管的生长和形成[16]。乳腺癌细胞中缺氧诱导因子-1(hypoxia inducible factor-1,HIF-1)激活可加快VEGF表达,进而加快细胞侵袭,诱导肿瘤血管形成[17-18]。本研究结果表明,下调MAC30可抑制乳腺癌细胞血管生成,从而抑制肿瘤生长,阻止疾病发展。MAC30虽然为IGF家族成员之一,但与IGF-1并不完全相同。MAC30在乳腺癌细胞中与IGF-1的表达水平一致。有研究发现,在乳腺癌细胞中,IGF-1的表达水平升高,并通过激活下游PI3K/AKT等,促进肿瘤细胞增殖、侵袭和转移等恶性生物学行为[19-20]。在乳腺癌细胞中IGF-1表达升高并参与肿瘤血管生成,因此推测下调MAC30可以抑制IGF-1活性,降低肿瘤组织中VEGF表达,减少血管形成数目,从而改善病情。本研究结果表明,下调MAC30后对ATP1A2活性具有调节作用,可以抑制乳腺癌的发生发展。
ATP1A2是一个编码钠钾泵α2亚基的基因,在细胞膜上扮演着重要的离子输送功能。现阶段,随着对ATP1A2基因研究的深入,其被认为与乳腺癌的发生发展存在一定关系。有研究表明,ATP1A2在乳腺癌组织中的表达水平显著高于正常组织,且ATP1A2高表达乳腺癌患者预后往往不佳[21]。ATP1A2对肿瘤细胞生物活性具有调控作用。有研究表明,上调ATP1A2可促进乳腺癌细胞的迁移和侵袭,而对其抑制后则能够减少乳腺癌细胞的迁移和侵袭[22]。本研究推测在乳腺癌中,MAC30的过表达可以调节SCr/PI3K/AKT通路的活性,从而促进肿瘤的增殖和侵袭,相反的抑制MAC30可能降低SCr/PI3K/AKT信号通路表达,从而发挥抗肿瘤血管生成的作用。本研究表明下调MAC30可以抑制ATP1A2、SCr、PI3K、AKT表达,进而抑制癌细胞生长,减少血管生成,加快焦亡。
综上所述,下调MAC30可抑制乳腺癌细胞增殖及血管生成,加快焦亡,其机制与抑制ATP1A2表达相关。但本研究未进行动物相关实验,关于MAC30调节ATP1A2的相关机制还需要进一步探讨。
