UPLC-MS/MS法测定利伐沙班中的基因毒性杂质
2023-06-07侯继鹏时晓燕张业华郝贵周张贵民鲁南制药集团股份有限公司国家手性制药工程技术研究中心山东临沂273400
侯继鹏,时晓燕,张业华,郝贵周,张贵民(鲁南制药集团股份有限公司,国家手性制药工程技术研究中心,山东 临沂 273400)
利伐沙班为一种高选择性抗凝药物,可直接抑制凝血因子Ⅹa,从而达到抗凝目的。临床上用于降低关节置换术后下肢静脉血栓风险,预防骨科大手术后深静脉血栓形成,提高非瓣膜性心房颤动患者抗凝治疗的临床疗效[1-5]。
利伐沙班的合成工艺主要使用三个起始物料,其中4-(4-氨基苯基)-3-吗啉酮为合成利伐沙班的中心前体,与化合物2-[(2S)-2-环氧乙烷基甲基]-1H-异吲哚-1,3-(2H)-二酮和5-氯噻吩-2-碳酰氯为原料合成利伐沙班[6]。这三个起始物料均含有基因毒性警示结构,具有潜在的基因毒性[7],它们及其副反应产物可能残留于利伐沙班。目前对于4-(4-氨基苯基)-3-吗啉酮及其副产物杂质、5-氯噻吩-2-碳酰氯及其副产物杂质、2-[(2S)-2-环氧乙烷基甲基]-1H-异吲哚-1,3-(2H)-二酮的研究均已有报道[8-14],但未见2-[(2S)-2-环氧乙烷基甲基]-1H-异吲哚-1,3-(2H)-二酮副产物杂质(S)-2-(2-羟基-氯丙基)-1H-异吲哚-1,3-二酮的相关研究。
本研究建立了超高效液相色谱-串联质谱法(UPLC-MS/MS)定量测定利伐沙班中的基因毒性杂质(S)-2-(2-羟基-氯丙基)-1H-异吲哚-1,3-二酮,根据人用药品注册技术要求国际协调会(ICH)M7和2020年版《中国药典》9306遗传毒性杂质控制指导原则相关规定[15-16],基于毒理学关注阈值(TTC)法,控制(S)-2-(2-羟基-氯丙基)-1H-异吲哚-1,3-二酮限度为50 μg·g-1,以保证利伐沙班的用药安全,现报道如下。
1 材料
1.1 仪器
Vanquish TSQ Altis型超高效液相色谱-质谱联用仪(美国Thermo公司);十万分之一电子天平MS105DU(瑞士Mettler Toledo公司);Milli-Q超纯水仪(美国Millipore公司)。
1.2 试药
利伐沙班原料药(山东新时代药业有限公司,含量>98.0%,批号:595190401、595190402、595190403、59171201、59171202、59171203);(S)-2-(2-羟基-氯丙基)-1H-异吲哚-1,3-二酮对照品(深圳斯坦德,含量:99.2%,批号:1894253R-YX-01);甲酸(质谱级,赛默飞世尔科技公司);乙腈为(质谱级,Merck公司);水(超纯水,美国Millipore 公司)。
2 方法与结果
2.1 色谱条件
UPLC系统:色谱柱 Thermo Hypersil Gold C18(2.1 mm×100 mm,3 μm);以0.1%甲酸为流动相 A,0.1%甲酸乙腈为流动相 B,梯度洗脱(0~10 min,20%~40%B;10~10.1 min,40%~20%B;10.1~15 min,20%B);流速 0.2 mL·min-1;柱温 25℃;进样量 20 μL。
质谱:离子源为电喷雾电离源(ESI),正离子模式采集;选择多监测模式(MRM),喷雾电压为3500 V,离子传输管温度为320℃;定量监测离子对m/z239.95→186.11,碰撞电压为16.30 V;定性监测离子对m/z239.95→221.97,碰撞电压为10.01 V。
2.2 溶液配制
2.2.1 对照品溶液的配制 精密称取(S)-2-(2-羟基-氯丙基)-1H-异吲哚-1,3-二酮对照品适量,加25%乙腈溶解并定量稀释制成150 ng·mL-1的溶液,作为对照品储备液;精密量取对照品储备液1 mL,置10 mL量瓶中,用25%乙腈稀释至刻度,摇匀,过滤,即得。
2.2.2 供试品溶液的配制 精密称取利伐沙班约15 mg,置50 mL量瓶中,依次加乙腈12.5 mL、水3 mL使溶解,用水稀释至刻度,摇匀,过滤,即得。
2.3 专属性
取空白溶液(25%乙腈)、对照品溶液和供试品溶液进样测定,结果见图1。(S)-2-(2-羟基-氯丙基)-1H-异吲哚-1,3-二酮的保留时间为6.667 min。空白溶液及供试品溶液不干扰杂质的测定。
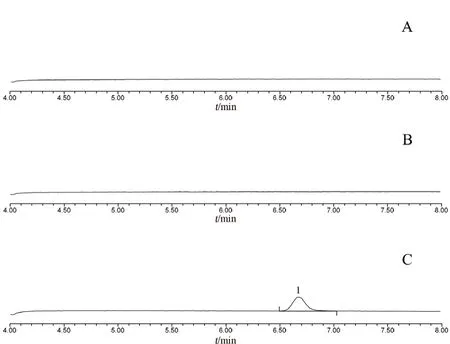
图1 专属性试验色谱图Fig 1 Specific test chromatogram
2.4 线性关系考察
精密量取对照品储备液适量,用25%乙腈分别稀释制成1.50、3.74、7.49、14.98、22.47、29.96 ng·mL-1的对照品溶液进样测定,记录色谱图。以质量浓度(C)为横坐标,峰面积(A)为纵坐标,进行线性回归,得标准曲线方程A=7.241×104C+1.778×104,r=0.9991,表明(S)-2-(2-羟基-氯丙基)-1H-异吲哚-1,3-二酮在1.50~29.96 ng·mL-1内与峰面积线性关系良好。
2.5 定量限和检测限
取对照品储备液,用25%乙腈逐级稀释至信噪比(S/N)约为10,测定定量限;用25%乙腈逐级稀释至S/N约为3,测定检测限。测得(S)-2-(2-羟基-氯丙基)-1H-异吲哚-1,3-二酮的定量限为1.50 ng·mL-1,检测限为0.45 ng·mL-1。
2.6 回收试验
精密称定利伐沙班15 mg,置50 mL量瓶中,依次加乙腈12.5 mL、水3 mL使溶解,共9份,各加入对照品储备液0.5、1、1.5 mL,用水稀释至刻度,摇匀,制成低、中、高浓度的溶液,每个浓度平行制备3份。依法测定,计算平均回收率,结果见表1,表明本方法准确度良好。
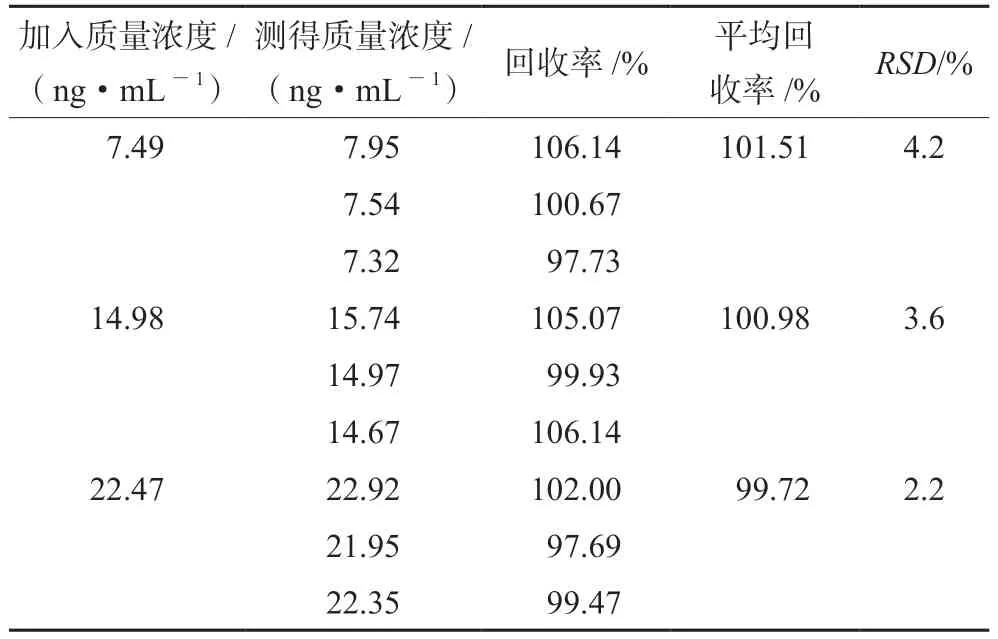
表1 回收试验结果Tab 1 Recovery
2.7 精密度、重复性试验
精密量取对照品储备液1 mL,置10 mL量瓶中,加25%乙腈稀释至刻度,摇匀,配制6份,进样测定,计算(S)-2-(2-羟基-氯丙基)-1H-异吲哚-1,3-二酮峰面积的RSD为0.63%(n=6),表明本方法精密度良好。
精密称取利伐沙班 15 mg,置50 mL量瓶中,依次加乙腈12.5 mL、水3 mL使溶解,加入对照品储备液1 mL,用水稀释至刻度,摇匀,配制6份,进样测定,结果(S)-2-(2-羟基-氯丙基)-1H-异吲哚-1,3-二酮含量的RSD为2.8%(n=6),表明本方法重复性良好。
2.8 稳定性试验
取新配制的对照品溶液,在0、3、6、9、12 h进样测定,记录色谱图。计算得(S)-2-(2-羟基-氯丙基)-1H-异吲哚-1,3-二酮峰面积的RSD为4.3%,表明对照品溶液于室温放置12 h内稳定。
2.9 样品测定
取6批利伐沙班原料药,按“2.2.2”项下方法配制供试品溶液。精密量取供试品和对照品溶液,分别进样测定。按外标法以峰面积计算6批利伐沙班中(S)-2-(2-羟基-氯丙基)-1H-异吲哚-1,3-二酮的含量,结果6批次利伐沙班中均未检出。
3 讨论
基因毒性杂质由于其毒性相对较高,引起了国内外药物工作者的高度重视,各药品管理机构发布了相应的指导原则,对基因毒性杂质推荐以TTC 1.5 μg·d-1来控制用药风险。基于TTC,根据利伐沙班产品说明书,以使用利伐沙班每日最大口服剂量30 mg·d-1计算,控制(S)-2-(2-羟基-氯丙基)-1H-异吲哚-1,3-二酮的限度须不超过50 μg·g-1。
方法摸索时,根据(S)-2-(2-羟基-氯丙基)-1H-异吲哚-1,3-二酮的结构,选择采用ESI离子源,正离子检测模式,使用三通连接注射泵、超高效液相色谱仪和质谱检测器,调整质谱的参数使杂质的响应达到最大;同时根据仪器优化的离子对,选择m/z239.95 → 186.11为定量离子对,m/z239.95 → 221.97为定性离子对,采用同时进行定性定量的方法提高检测方法的准确度,防止假阳性结果的产生。
应用TTC法计算得到杂质的限度较低,当质谱常规进样量为5 μL时发现,杂质的响应无法达到检测限度要求,故提高进样量至20 μL,使方法满足限度检测要求;但此时系统进样体积较大,而UPLC系统的管路及色谱柱较细,杂质产生较强的溶剂效应,峰形较差。根据流动相中有机相的比例,测试25%、30%、40%乙腈为稀释液发现,使用25%和30%乙腈为稀释液时(S)-2-(2-羟基-氯丙基)-1H-异吲哚-1,3-二酮峰形较好,使用40%乙腈峰形变差,最终选择25%乙腈为稀释液。
利伐沙班在水中溶解性较差,直接使用25%乙腈配制质量浓度为0.3 mg·mL-1的利伐沙班时,供试品无法完全溶解;经摸索发现可以先加部分乙腈和水让利伐沙班原料溶解,然后再加水进行定容,用25%乙腈作为稀释液的配制思路进行供试品的配制。通过加标验证该配制方法杂质回收率较好,解决了原料无法完全溶解的问题。
