重构人类演化中族群融合与基因交流历史的方法
2023-05-30张瑞徐书华
张瑞 徐书华
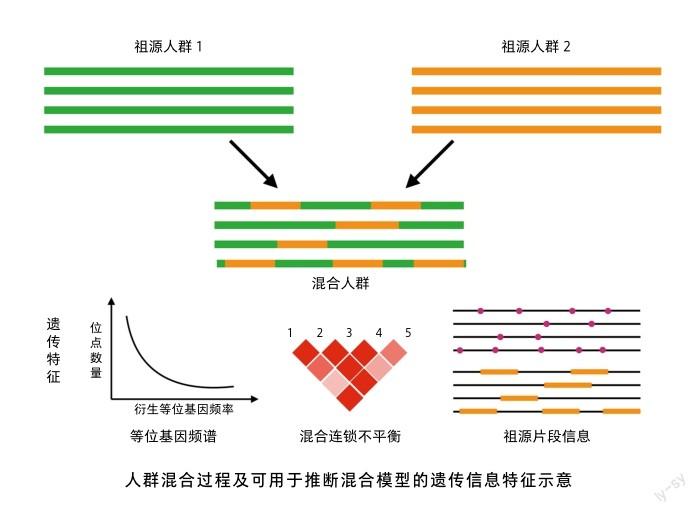
遗传混合是人类演化的重要驱动力之一。实际上,世界上大部分人群都是混合人群,并且其混合过程随着时间的推移越来越复杂。其中最典型的混合人群有非裔美国人(African American)、拉丁裔美国人(Latino American),以及我国西北地区的维吾尔族和哈萨克族人群等。准确而高效的统计模型与算法工具,可以为解析混合人群的祖源构成、重构混合历史模型,以及阐明人类遗传多样性的产生和演化机制,提供强有力的理论支持与技术支撑。下面主要从统计方法和算法工具的角度,对人群混合历史研究中的四个关键科学问题进行系统介绍:全局祖源推断(global ancestry inference),局部祖源推断(local ancestry inference),现代人类混合历史重构(modern human admixture history modeling)和远古人类与现代人类基因交流历史重构(archaic introgression history modeling)。
全局祖源推断
全局祖源推断是指在混合个体的全基因组水平上,估计各祖源人群对于混合人群的贡献比例,依据其理论框架不同可大致分为非参数方法和基于模型的方法。在非参数方法中,较为典型且常用的为主成分分析。该方法通过对输入数据进行降维,提取能最大化描述输入数据方差且线性无关的一些特征,根据不同特征所能解释数据方差程度的大小排序,再依次将其命名为第一主成分、第二主成分等。利用主成分分析结果,可以近似地用低维数据表示原始输入数据,并进而研究其内部数据结构,达到对高维数据进行降维的效果。类比到全局祖源推断中,人类全基因组大约有30亿个碱基,如此高维的数据难以直接反映出人群内部遗传结构。通过对人群原始遗传数据进行主成分分析,再分别以第一主成分和第二主成分为横纵轴,可以看到所有的个体数据被投影到一个平面内,而相距较近的个体表明其遗传关系也更为接近。此外,若对世界范围内各大洲的基因组数据进行主成分分析,不难发现在地理位置上相对较近的人群,在主成分分析结果图上也会呈现出相对聚集的模式,这体现了世界范围内人群地理位置信息可以与其遗传关系存在一定程度上的对应。然而,基于主成分分析的结果仅能大致判断哪些人群或个体在遗传关系上较为接近,而无法进一步估计混合比例等参数。
接下来,就是基于模型进行全局祖源推断。此类方法中使用较多的工具是普里查德(J. K.Pritchard)等在2000年提出的STRUCTURE[1],以及亚历山大(D. H. Alexander)等在2009年提出的ADMIXTURE[2]。这两类工具均是在给定祖源成分数量(记为K)的前提下,对输入人群进行聚类并产生K个不同的簇(cluster)。其中,STRUCTURE基于贝叶斯方法,通过马尔可夫链蒙特卡罗(Markov Chain Monte-Carlo, MCMC)方法对后验分布进行采样,进而推测输入数据中存在的群体遗传结构。ADMIXTURE软件借鉴了STRUCTURE的似然模型,并利用极大似然方法来估计群体遗传结构。该方法在不显著影响准确度的前提下,运行速度相比于STRUCTURE提升数十倍。这两类方法均可以在全基因組水平上估计祖源人群对混合人群的贡献比例。
局部祖源推断
不同的混合过程可能会呈现出相似的全局祖源模式。有研究团队发现,存在地理隔离的不同人群与进行长距离历史迁徙的人群相比,所呈现的主成分分析结果十分接近[3]。为进一步了解混合人群的遗传结构,需要利用局部祖源推断方法。局部祖源推断是从混合人群的单倍体水平上,解析其基因组中每个位点或者每个片段最有可能来自的祖源人群,这也可以理解成一个给混合个体基因组进行标注的过程。相比于全局祖源推断,局部祖源推断对于祖源成分的解析会更加精细和具体化。目前较常见的局部祖源推断方法主要基于隐马尔可夫模型,它将混合个体在某个位点上的等位基因看作是观测状态,并将位点的祖先来源看作是隐状态。基于隐马尔可夫模型中所定义的初始状态概率向量(即在初始时刻不同隐状态的概率),状态转移概率矩阵(即此刻某一隐状态跳转到下一时刻某一隐状态的概率),以及发射概率矩阵(即每一隐状态生成不同观测的概率),可以利用当前时刻所观测到的信息,以及此前时刻的隐状态推测当前时刻最有可能的隐状态。类比到局部祖源推断的问题中,即结合当前位点所观测到的等位基因信息,以及此前位点的祖先来源状态,利用隐马尔可夫模型推测该位点最有可能来源的祖源人群状态。
李(N. Li)和斯蒂芬斯(M. Stephens)最早于2003年提出基于隐马尔可夫模型结合局部祖源信息和重组率估计的理论框架。此后,该模型框架被不断应用于局部祖源推断方法的开发中,并取得了较好成果。此外,条件随机场(conditional random field, CRF)模型,支持向量机(support vector machine, SVM)模型,以及随机森林(random forest, RF)模型等也被应用于局部祖源推断方法的开发中。这些方法中,较典型的有以下三个工具:迈尔斯(S. Myers)研究组在2009年开发的HAPMIX[4],布斯塔曼特(C. D. Bustamante)研究组在2013年开发的RFMix[5],以及布卢姆(M. G. B. Blum)研究组在2018年开发的Loter[6]。它们所基于的理论框架和算法模型不尽相同,因此其所适用的群体遗传数据分析场景也存在一定差异:HAPMIX对于推断两祖源人群混合模式(two-way admixture)下的局部祖源十分准确;RFMix主要适用于推断混合时间较为近期的混合群体,例如非裔美国人和拉丁裔美国人等;Loter更适用于推断混合时间较为久远的混合群体,大致为距今3000年前发生的混合事件。
现代人类混合历史重构
混合历史的构建主要包括混合模式的推断,混合时间,以及混合比例的估计。目前人类基因组中用于推断人群混合历史的遗传特征主要分为以下三种:等位基因频谱(allele frequency spectrum),混合连锁不平衡(admixture linkage disequilibrium, ALD)以及祖源片段信息(ancestral tracts)。
等位基因频谱描述的是人群在给定位点上的等位基因频率的分布情况,其能很好地刻画基因突变和遗传漂变的信息。等位基因频谱的变化在一定程度上可反映人群历史信息,且等位基因频率的计算方式也较为简单,可以直接依据基因型数据计算,无需大量的算法迭代过程,所以早期用于推断人群混合历史的方法多基于这一遗传信息。其中,比较有代表性的是赖克(D. Reich)团队在2009年开发的ADMIXTOOLS工具[7],该方法提出一系列统计量,如D统计量、F3统计量等,并基于不同人群的等位基因共享情况,借助系统发生树拓扑结构来刻画不同人群之间的基因流动与遗传混合情况。
连锁不平衡(linkage disequilibrium, LD)描述了群体水平位点之间的非随机的相关性,即在某一群体中某两个位点之间的相关性明显高于随机情况的现象。LD的产生与自然选择、遗传漂变、群体结构、有效群体大小均有关系。而ALD指的是混合过程中,祖源人群内部的特异性位点在混合人群基因组中更倾向于连锁在一起的现象。祖源人群分歧时间越久,各个人群内部积累的特异性位点也会随之增多,在形成混合人群时,ALD也会越强。但是在混合事件发生之后,随着混合时间的增加,ALD会以一定的速率指数衰减。所以,结合混合事件发生时的LD以及当前混合人群中的LD信息,可以推断混合历史。以LD推测混合历史的思路最先被穆尔贾尼(P. Moorjani)及其合作者应用,随后被罗(P. Loh)与皮克雷尔(J. K. Pickrell)等人进一步改进,并于2013年和2014年分别提出了ALDER以及MALDER工具。
祖源片段信息是混合个体单倍型水平上每个位点或者片段的祖先来源,这一信息的提取可以借助前面介绍的局部祖源推断方法。祖源片段信息大致可分为祖先间跳转速率(ancestral switches probability)和祖源片段长度分布(ancestral tracts length distribution)。前者描述了混合人群基因组中,不同祖先来源的片段之间发生转换的概率。当两个群体之间的混合时间越长,随着重组事件的积累,对应祖先间片段跳转的次数就越多。而后者描述的是混合人群基因组中,来自某一祖源人群的片段长度分布情况。在群体遗传学中,重组事件的发生可以用泊松过程来刻画,因此,在混合过程中由于重组被打碎的祖源片段长度服从指数分布。随着混合时间的增加,重组事件积累的次数变多,祖源片段也会越来越碎。另外,来自某一祖源片段的总长度也可以反映该祖源人群的遗传物质在混合群体中所占的比例。祖源片段长度信息最早在2009年被普尔(J. E. Pool)和尼尔森(R. Nielsen)应用于混合历史的推断中。此后,自2018年起,笔者研究团队也基于此遗传信息开发了MultiWaver系列软件[8],该软件可以在不同混合模式下分别进行参数估计,并依据不同混合模型似然值大小选择最优混合模型。
以上提及的方法均为常染色体水平上的混合历史推断,然而,很多混合事件都带有性别偏向性,即特定祖源的男女遗传贡献存在差异,如非裔美国人为学界熟知的带有性别偏向性混合模式的人群。传统研究性别偏向性混合的方法多从单系遗传的Y染色体非重组区域或者线粒体DNA出发,通过对不同单倍型进行分类,从而判断对应祖源在混合发生时带有的性别偏向的方向。此外,目前也有较多的研究通过比较常染色体与X染色体在全局水平上混合比例的差异,来判断是否存在性别偏向性,并估计具体的偏离程度,其中全局水平上常染色体和X染色体混合比例的估计,多借助于前面提及的全局祖源推断方法。当某一祖源贡献的常染色体混合比例大于X染色体时,则该祖源人群混合偏向为男性主导(malebiased),反之为女性主导(female-biased)。筆者研究团队于2022年基于MultiWaver系列软件模型框架开发出了MultiWaverX工具[9]。该工具可用于重构精细尺度的性别偏向性混合历史,即精确量化混合过程中,每一祖源人群每次混合事件带有的性别偏向性方向以及具体偏离程度。值得一提的是,研究团队发现一种较为特殊的性别偏向性混合模式:抵消模型,即混合过程中两次或多次携带不同方向的性别偏向性混合事件得以抵消,最终呈现出无性别偏向的混合模式,且这类混合模式不能通过目前已有的其他方法进行解析。
远古人类与现代人类基因交流历史重构
上文提及的均为不同现代人群即智人(Homo sapiens)后代之间的混合,除了早期智人之外,地球上也曾存在过其他古人类,目前考古学研究较多的为尼安德特人(Homo neanderthalensis)和丹尼索瓦人(Denisova hominin),他们在距今4万—3万年前走向灭绝。这些古人类与现代人类祖先在时间和空间上都存在着一定的交集,目前的研究表明,早期智人曾与尼安德特人和丹尼索瓦人有过多次接触。受限于目前可获取的古人类基因组信息,以下关于古人类的讨论均指尼尔德特人或者丹尼索瓦人。由于现代人类基因组中由古人类渗入的片段大都受到负向选择或者存在遗传漂变等因素,其留存下来的古人类渗入片段比例相对较小,为1%~3%。因此,混合时间较久、留存比例较小等因素,给检测古人类基因渗入以及重构古人类与现代人类基因交流历史带来了巨大挑战。
远古人类对现代人类祖先的基因渗入,可以视为一种极端情况下的人群混合,因此一些用于重构现代人类混合历史的遗传特征,例如等位基因频率与连锁不平衡信息等,也同样适用于古人类与现代人类混合历史推断。古人类与现代人类基因交流历史研究一般可分为以下两个部分:一是检测现代人类基因组中古人类渗入比例以及定位古人类渗入片段;二是构建古人类与现代人类混合历史,其中包括确定混合次数以及估计每次的混合时间与混合比例。
对于第一部分,依据所参考的基因组信息不同,将已有的方法分成三类。第一类是仅利用外群(outgroup)的基因组信息,外群是指没有古人类基因渗入的人群。这类方法一般通过与外群基因组信息进行比较,筛选掉那些不太可能为古人类渗入的位点。这一般不需要借助古人类基因组序列,因此该方法也可用于检测由其他未知古人类渗入的片段。在外群选取上,目前研究表明非洲人群基因组中携带着的由古人类渗入的片段较少或者几乎没有,故多以非洲人群作为外群。这类方法的代表工作是阿基(J. M. Akey)研究组在2016年开发的S* [10]。该方法借助于检测现代人群中显著强连锁的片段来寻找古人渗入片段。当两祖源人群之间的分歧时间较久时,他们形成的混合人群会呈现出较强的LD模式。古人与现代人之间的分歧时间可以追溯到70万年以前,他们各自携带的人群特异性位点也较多,所以当古人对现代人基因渗入时,在现代人基因组上为古人特有等位基因之间的LD会特别强。第二类方法则同时利用外群和古人类基因组信息,具体为在推断古人渗入片段时考虑在外群人群中频率较低、但与古人基因组存在一定匹配性的片段。目前这一类方法有基于等位基因频率的D统计量和F4 ratio统计量,通过判断现代人群与古人类之间的衍生等位基因的共享情况来估计古人类渗入比例。这两个统计量也包含于上文提及的ADMIXTOOLS工具中。此外,笔者研究团队也于2021年提出ArchaicSeeker 2.0方法,其能有效利用单倍型序列信息并基于隐马尔可夫模型来检测现代人基因组中由古人类渗入的序列片段[11]。第三类方法则仅利用古人类基因组信息。部分学者认为找到完全无古人类渗入的现代人群是相對困难的,若利用带有古人类渗入的群体作为外群,必然会给分析过程引入一定的偏差,所以这类方法只用于借助古人类基因组信息推断现代人群中古人类渗入的片段。目前这类方法中较为主流的工具为阿基研究组在2020年开发的IBDmix软件[12],其通过检测现代人与古人类基因组之间的祖先同源片段(identity by descent, IBD)来推测某一位点或者区域是否为古人类渗入状态,其中IBD片段为两个个体中状态相同且来自同一个共同祖先的片段。
相比第一部分中估计古人类渗入比例或者定位渗入片段,第二部分重构古人类与现代人类混合历史模型更为复杂。传统方法大都借助大量的计算机模拟,以期找到与目标人群最为相似的混合方式。前文提到的笔者研究团队开发的ArchaicSeeker 2.0方法,在不依赖海量计算机模拟的前提下,可进一步利用古人类渗入片段长度分布信息有效重构极为复杂的遗传渐渗历史。
结 语
综上所述,运用统计方法、算法工具以及人类全基因组数据,研究人员解析了世界范围内现代人类迁徙与混合的历史进程,同时也在不断探索更久远的时间尺度内,远古人类与现代人类祖先之间复杂多样的混合历史结构。
在现代人类混合历史研究中,除了人们较为熟悉的非裔美国人混合模式之外,祖源成分更多,且混合模式更复杂的人群历史也逐渐被解析出来。例如,笔者团队发现中国新疆维吾尔族人群基因组中有源自东亚、南亚、西欧和西伯利亚等四个区域人群的遗传成分,且呈现出“混合之混合”的复杂模式,即东亚与西伯利亚人群先发生混合,西欧与南亚人群再发生混合,之后,先前产生的两个混合人群之间发生基因交流,形成当今的新疆维吾尔族[13]。在古人类与现代人类的基因交流研究中,笔者研究团队发现,现代人类与远古人类间存在多次基因交流。早期走出非洲的现代人类祖先,在距今11.9万~9.4万年前,在中东、南亚附近与丹尼索瓦人发生了第一次接触和基因交流。而后,他们继续向东前进,一部分留在南亚,一部分向北到达东亚南部,并分别与当地的丹尼索瓦人发生遗传交融。而后,一支现代人祖先继续向东南迁徙,在距今6.2万~6.4万年前,穿过华莱士线到达大洋洲,并与先前到达的丹尼索瓦人融合。而近期走出非洲的现代人类祖先,在距今5.9万~4.8万年前,在中东地区与尼安德特人发生第一次接触和基因交流。之后,他们分别迁徙至欧洲、南亚,以及东亚,与各地的尼安德特人发生了第二次族群融合[11]。
随着基因组测序技术的改进和成本的下降,可以更容易地获取世界范围内现代人类与古人类的高质量基因组测序数据,这一定程度上也在挑战现有统计方法和算法工具的适用场景。从以上算法工具的提出时间来看,对于现代人类混合历史研究的方法均在2000年之后,而人类基因组序列草图是在2000年6月初步完成的。此后,一系列国际基因组计划也不断开展,其中包括2007年国际人类基因组单体型图(HapMap)计划和2012年的千人基因组计划等。对于古人类与现代人类混合历史研究的方法多于2014年之后提出,而第一个丹尼索瓦人和第一个尼安德特人的全基因组高倍测序序列正是分别于2012年和2014年完成。随着数据挖掘、机器学习以及深度学习等领域的发展,更高效的统计模型与算法工具也会进一步发展,并被应用到群体遗传学的研究中。理论模型、计算方法与遗传数据三者相辅相成,为深入探究现代人类和远古人类在宏大时空框架下的分化、融合与适应性演化历史提供了更多可能,并为进一步解析人类起源与演化中更深层次的基础理论问题提供了新的视角。
[1]Pritchard J K, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics, 2000, 155: 945-959.
[2]Alexander D H, J. Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Research, 2009, 19: 1655-1664.
[3]Novembre J, Johnson T, Bryc K, et al. Genes mirror geography within Europe. Nature, 2008, 456: 98-101.
[4]Price A L, Tandon A, Patterson N, et al. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genetics, 2009, 5: e1000519.
[5]Maples B K, Gravel S, Kenny E E, et al. RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. American Journal of Human Genetics, 2013, 93: 278-288.
[6]Dias-Alves T, Mairal J, Blum M G.B. Loter: A software package to infer local ancestry for a wide range of species. Molecular Biology and Evolution, 2018, 35: 2318-2326.
[7]Reich D, Thangaraj K, Patterson N, et al. Reconstructing Indian population history. Nature, 2009, 461: 489-494.
[8]Ni X, Yuan K, Liu C, et al. MultiWaver 2.0: modeling discrete and continuous gene flow to reconstruct complex population admixtures. European Journal Human Genetics, 2019, 27: 133-139.
[9]Zhang R, Ni X, Yuan K, et al. MultiWaverX: modeling latent sexbiased admixture history. Briefings in Bioinformatics, 2022, 1-12.
[10]Vernot B, Tucci S, Kelso J, et al. Excavating Neanderthal and Denisovan DNA from the genomes of Melanesian individuals. Science, 2016, 352: 235-239.
[11]Yuan K, Ni X, Liu C, et al. Refining models of archaic admixture in Eurasia with ArchaicSeeker 2.0. Nature Communications, 2021, 12: 6232.
[12]Chen L, Wolf A B, Fu W, et al. Identifying and interpreting apparent Neanderthal ancestry in African individuals. Cell, 2020,180: 677-687.
[13]Feng, Q, Lu Y, Ni X, et al. Genetic history of Xinjiangs Uyghurs suggests Bronze Age multiple-Way contacts in Eurasia. Molecular Biology and Evolution, 2017, 34: 2572-2582.
關键词:混合人群 祖源推断 基因交流 现代人类 远古人类 ■
