咖啡果皮与不同来源可溶性膳食纤维结构及性质比较研究
2023-05-30朱珂董文江程金焕胡荣锁何红艳陈小爱龙宇宙黄家雄
朱珂 董文江 程金焕 胡荣锁 何红艳 陈小爱 龙宇宙 黄家雄
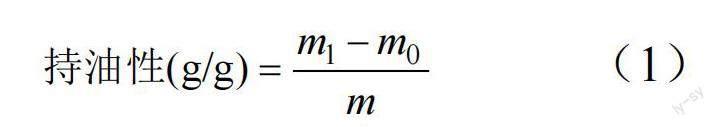

关键词:咖啡果皮;可溶性膳食纤维;微观结构;功能特性
中图分类号:TS273;TS209 文献标识码:A
咖啡为茜草科常绿灌木或小乔木,与可可、茶并称为世界三大饮料,原产于埃塞俄比亚。我国咖啡主要种植区为海南和云南,其中海南万宁的“兴隆咖啡”为中粒种罗布斯塔咖啡,具有醇厚度高、焦糖香味浓郁等特点,国家质检总局于2007 年授予“兴隆咖啡”国家地理标志保护产品,并于2021 年获得“中欧100+100”地理标志互认互保产品。
据农业农村部统计,2019 年我国咖啡总产量为1.379×105 t,居全球第13 位,随着咖啡产量的持续增长,咖啡加工过程中产生的副产物也在逐年增加,包括咖啡果壳、咖啡果皮、银皮、咖啡渣等,这些副产物大多被作为垃圾直接扔掉[1],不仅造成资源浪费,还会造成环境污染。研究表明,这些副产物富含生物活性物质(咖啡因、绿原酸、葫芦巴碱等),在制药、食品、化妆品行业中可能具有潜在应用价值[2]。目前,国内外对于咖啡副产物的研究主要集中在生物燃料、饲料、生物吸附剂、抗氧化剂以及聚合物复合材料的制造等方面[3]。
咖啡果皮是湿法加工过程中产生的副产物,含35%~80%的碳水化合物,总膳食纤维含量可达70%左右,还含有4%~12%的蛋白质、0.5%~3%的脂类、3%~10%的矿物质、1%~9%的单宁以及约1%的咖啡因,具有较高的利用价值[4]。胡荣锁等[5]采用响应面法优化了酶法提取咖啡果皮可溶性膳食纤维的条件,王彦兵等[6]通过响应面法优化了超声辅助提取咖啡果皮多酚的提取条件,确定了最佳提取条件。ESQUIVEL 等[7]对比了不同提取方法从阿拉比卡与罗布斯塔2 个品种的咖啡果皮中提取生物活性成分,研究其抗氧化性的差异,结果表明,不同提取方法所得的活性成分含量及抗氧化性存在差异;DONG 等[8]研究了5 种不同提取方法对咖啡果皮可溶性膳食纤维结构及性质的影响,结果表明剪切乳化辅助酶法的提取率最高,且所得样品具有较好的功能特性。
膳食纤维(dietary fiber, DF)是一类不易被人体消化酶消化的碳水化合物,包括纤维素、半纤维素、木质素、果胶等。根据其溶解性的不同分为可溶性膳食纤维(soluble dietary fiber, SDF)和不溶性膳食纤维(insoluble dietary fiber, IDF)两大类。SDF 主要有果胶、树胶等亲水性胶体物质及部分半纤维素,具有重要的生理功能[9],如降血糖、降血脂、调节血压等[10]。膳食纤维的来源不同,其理化特性、结构均有差异,目前,大量的果蔬、谷物及其副产物是膳食纤维的主要来源,如龙芳[11]研究了不同提取方式对芹菜可溶性膳食纤维结构和理化特性的影响。咖啡果皮可溶性膳食纤维作为一种功能性碳水化合物,其与常见的大宗豆类、果蔬中可溶性膳食纤维组成及结构存在哪些异同点尚无报道,解析咖啡果皮相对其他来源可溶性膳食纤维的优势和特色之处,对于咖啡果皮的深度开发利用具有重要的指导意义。
本研究通过对大豆、菊粉、大枣、燕麦、芹菜5 种市售可溶性膳食纤维与传统水提法制备的咖啡果皮可溶性膳食纤维在粒径分布、单糖组成、微观结构、熱稳定性及功能特性等进行比较,探究不同来源可溶性膳食纤维在单糖组成、微观结构、理化性质及功能特性等方面的异同点,分析咖啡果皮可溶性膳食纤维对比其他样品的优异性质,为咖啡果皮可溶性膳食纤维的开发利用奠定理论基础,为咖啡果皮副产物的高值化利用提供理论依据。
1 材料与方法
1.1 材料
1.1.1 材料与试剂 咖啡鲜果(全红果)采摘于中国热带农业科学院香料饮料研究所咖啡试验基地。乙醇(西陇科学有限公司);氢氧化钠(深圳特镨超纯材料科技有限公司);冰乙酸、邻苯二甲醛、硝酸银、溴化钾、NaNO2、盐酸萘乙二胺、对氨基苯磺酸等均为分析纯,购自上海阿拉丁公司;葡萄糖、阿拉伯糖、果糖、鼠李糖、木糖、甘露糖、岩藻糖、半乳糖醛酸和葡萄糖醛酸均为色谱纯,购自上海阿拉丁公司。
1.1.2 仪器与设备 Nicolet 6700 傅里叶变换红外光谱仪(美国赛默飞世尔科技公司);PhenomProx 台式显微能谱一体机(荷兰复纳科学仪器有限公司);Avanti JXN-26 高速冷冻离心机(美国贝克曼库尔特有限公司);ICS-5000+型离子交换色谱(美国赛默飞世尔科技公司);Mastersize 3000激光粒度分析仪(英国马尔文仪器有限公司);Specord 250 Plus 紫外可见分光光度仪(德国耶拿分析仪器股份公司);DSC2500 差示扫描量热仪(美国TA 仪器公司);帕纳科XPertPRO(荷兰帕纳科公司)。
1.2 方法
1.2.1 咖啡果皮可溶性膳食纤维的制备 (1)咖啡果皮预处理。湿法加工工艺流程:采收→鲜果浮选→机械脱皮脱胶→水洗→干燥→咖啡豆。将湿法加工得到的咖啡果皮于40℃热风干燥至水分含量为11%左右,高速万能粉碎机粉碎后过60目筛,置于密封袋中,4℃保存备用。
(2)咖啡果皮可溶性膳食纤维的提取。参考褚盼盼等[12]的方法并略作修改,采用传统水提法提取可溶性膳食纤维,称取200 g 咖啡果皮粉至烧杯中,按照料液比1∶40(g/mL)加入去离子水,90℃水浴60 min 后滤去残渣,取上清液50℃下旋转蒸发至原体积的1/6,加入4 倍体积的乙醇溶液醇沉1 h,抽滤后取滤渣,真空冷冻干燥后得咖啡果皮可溶性膳食纤维,装入密封袋于干燥器中保存备用。
1.2.2 粒径分析 采用Mastersize 3000 激光粒度分析仪干法测定可溶性膳食纤维粉末的粒径分布情况,取样品0.05 g 置于料斗中进行测量,样品粒径以D10、D50、D90 和D99 来表示,分别代表样品颗粒累积分布为10%、50%、90%和99%时对应的粒径大小。
1.2.3 单糖组成分析 参考LI 等[13]的方法,准确称量20 mg 样品于安瓿管中,加入2.0 mL2 mol/L 的三氟乙酸溶液,真空封管后于110℃下加热水解3 h,旋干三氟乙酸后,加入1.0 mL 甲醇溶液洗涤、旋干,重复3 次,加入去离子水稀释配置成1.0 mg/mL 样品试液。
采用高效阴离子交换色谱- 脉冲安培法(HP-SEC)测定10 种单糖标准品(葡萄糖-Glu、阿拉伯糖-Ara、木糖-Xyl、半乳糖-Gal、半乳糖醛酸-GalA、岩藻糖-Fuc、鼠李糖-Rha、果糖-Fru、半乳糖醛酸-Gala、葡萄糖醛酸-GlcA)的组成及含量。仪器配置安培检测器,利用Diobex CarboPacPA20(150 mm×3 mm, 6.5 μm)高效阴离子色谱进行分离,配备CarboPac PG20 保护柱(30 mm×3 mm,6.5 μm)。流动相:A 相,去离子水;B 相,200 mmolNaOH 溶液;C 相,1 mol/L NaAC/25 mmol NaOH。流速为0.5 mL/min,进样量为25 μL。洗脱条件:0~5 min,9% B;5~14 min,4% B;14~24 min,4% B、5% C;24~24.1 min,4% B、20% C;24.1~40 min,100% B。结果以g/100 g 表示。
1.2.4 结构表征 (1)傅里叶变换红外光谱(FT-IR)。参考GU 等[14]的方法,准确称量干燥的5.0 mg 可溶性膳食纤维粉于玛瑙研钵中,加入500 mg(1∶100)干燥的光谱级KBr 在红外灯照射下研磨至极细,混合均匀压片,将压好的透明薄片置于样品槽中进行扫描分析,扫描次数64次,分辨率为4 cm–1,扫描范围为400~4000 cm–1。
(2)扫描电镜(SEM)。扫描电镜可以对样品微区形貌、结构及成分进行观察和分析。参考MA等[15]的方法,在15 kV 条件下用扫描电子顯微镜观察,将微量的干燥样品使用导电胶固定于样品台上,在真空条件下喷金后置于电子显微镜下扫描,观察样品放大200 倍时的微观结构。
(3)X-射线衍射(XRD)。X-射线衍射是通过X-射线在晶体中所产生的衍射现象反映样品的结晶特性及结晶度。参考WEN 等[16]的方法略有修改,采用XPert PRO 对可溶性膳食纤维样品进行XRD 测定。测定参数:测试靶材为铜靶;扫描速度为2°/min;扫描范围为5~70°。
(4)差示扫描量热法(DSC)。采用DSC2500差示扫描量热仪进行可溶性膳食纤维的热稳定分析。称取少量样品,置于DSC 铝盘中压片密封,置于室温平衡后,以10℃/min 的速度从35℃升至200℃。采用空铝盘作空白对照。
1.2.5 可溶性膳食纤维的理化性质测定 (1)持油性。参考ZHANG 等[17]的方法,准确称量0.5 g可溶性膳食纤维样品(质量为m)置于10 mL 离心管中,质量为m0,加入5 mL 花生油混合均匀,4℃下静置1 h,4800 r/min 离心10 min,弃掉上层油脂,质量为m1,持油性公式如下:
式中,m0、m1 分别为离心前后可溶性膳食纤维粉质量,m 为初始可溶性膳食纤维粉质量。
(2)溶解性。准确称量0.1 g 可溶性膳食纤维样品,质量为m,加入5 mL 蒸馏水于90℃水浴1 h,冷却至室温后4200 r/min 离心10 min,上清液置于已称重培养皿(m0)中烘至恒重后,称重(m1),溶解性公式如下:
式中,m0、m1 分别为烘干前后盛放可溶性膳食纤维培养皿的质量,m为初始可溶性膳食纤维粉质量。
1.2.6 功能特性测定 (1)胆固醇吸附能力。参考罗白玲等[18]的方法略作修改,采用邻苯二甲醛比色法制作标准曲线:分别取0、0.1、0.2、0.3、0.4、0.5 mL 0.1 mg/mL 胆固醇标准溶液,冰乙酸对应补足至0.5 mL,混匀后加入0.5 mL 邻苯二甲醛(OPA)试剂(50 mg 邻苯二甲醛溶于冰乙酸,定容至100 mL)和4.0 mL 混合酸(浓硫酸和冰乙酸等体积混合),充分混匀后室温下静置10 min,在550 nm 处测定吸光度,绘制标准曲线。
取2 个新鲜鸡蛋,分离蛋黄与蛋清,在蛋黄中加入9 倍体积的蒸馏水用均质机打成均匀的乳液。取0.5 g 样品加入25 mL 搅打均匀的蛋黄乳液,调节pH 至7.0,37℃恒温震荡2 h,4800 r/min离心10 min,取1.0 mL 上清液,去离子水稀释20 倍后测定吸光度。
胆固醇吸附量(mg/g)=(m1-m2)/m (3)
式中,m1 为吸附前蛋黄乳液中胆固醇质量,mg;m2为吸附后上清液中胆固醇质量,mg;m 为样品质量,g。
(2)胆酸盐吸附能力。参考HUANG 等[19]的方法稍作修改。分别配置0.2、0.4、0.6、0.8、1.0 mg/mL 的胆酸钠水溶液,吸取不同浓度胆酸钠水溶液1.0 mL 于比色管中,加入6.0 mL 45%硫酸和1.0 mL 0.3%糠醛,混合均匀后放入65℃恒温水浴锅中加热30 min,然后取出冷却至室温,取部分反应后的溶液在620 nm 处测定吸光度,绘制标准曲线。
准确称量样品0.25 g 于离心管中,加入含0.1 g 胆酸钠的0.15 mol/L 氯化钠溶液25.0 mL,调节pH=7,在37℃下恒温震荡2 h,4800 r/min离心10 min,取1.0 mL 上清液按上述方法测定吸光度。
胆酸盐吸附量(mg/g)=(m1-m2)/m (4)
式中,m1 为吸附前胆酸钠质量,mg;m2为吸附后上清液中胆酸钠质量,mg;m 为样品质量,g。
(3)葡萄糖吸附能力。参考阮传英等[20]的方法略作修改,采用二硝基水杨酸(DNS)法制作标准曲线,配制0.5 mg/mL 的葡萄糖标准液,分别吸取0、0.2、0.4、0.6、0.8、1.0 mL 标准液于比色管中,加入去离子水至3.0 mL,加入DNS试剂2.0 mL,在沸水浴中反应10 min,空白管调零,在540 nm 处测定吸光度,绘制标准曲线。
取SDF 样品0.1 g 与25 mL 50 mmol/L 的葡萄糖标准液混合,在37℃水浴中震荡2 h 后,4800 r/min离心10 min,取上清液并按上述方法测定吸光度。由于样品本身含有葡萄糖,需另准备一组不加葡萄糖的样品作为阴性对照。
葡萄糖吸附量(mg/g)=(m1-m2)/m (5)
式中,m1为吸附前葡萄糖质量,mg;m2为吸附后上清液中葡萄糖质量,mg;m 为样品质量,g。
(4)亚硝酸根离子吸附能力。参考DONG 等[8]的方法略作修改,配制0.1 mg/mL 的亚硝酸钠标准液,分别取0.1、0.2、0.3、0.4、0.5 mL 标准液于比色管中,加水至2.0 mL,加入2.0 mL 对氨基苯磺酸溶液(4.0 g/L),再加入1.0 mL 盐酸萘乙二胺(2.0 g/L),室溫下静置15 min 后在538 nm处测量吸光度,绘制标准曲线。
取SDF 样品0.25 g 与25.0 mL 标准液混合均匀,分别调节pH 为2(模拟胃酸环境)和pH 为7(模拟小肠环境),在37℃水浴中震荡2 h,4800 r/min 离心10 min,取上清液0.5 mL 按上述方法测定吸光度。
亚硝酸盐吸附量(mg/g)=(m1-m2)/m (6)
式中,m1 为吸附前亚硝酸钠质量,mg;m2 为吸附后上清液中亚硝酸钠质量,mg;m 为样品质量,g。
1.3 数据处理
采用Origin 2021(Northampton, MA, USA)软件绘图,采用SPSS 20.0(IBM Corporation, NewYork, NY)软件中Duncan 多重比较进行一元方差分析并比较样品间指标的差异性。每个样品平行测定3 次,结果以平均值±标准偏差形式表示。
2 结果与分析
2.1 粒径分析粒径大小与可溶性膳食纤维理化性质、功能特性密切相关。咖啡果皮、大豆、菊粉、大枣、燕麦和芹菜可溶性膳食纤维的中值粒径D50分别为131.49、57.17、55.33、64.43、50.17、39.33 μm,不同样品间粒径大小存在显著差异(表1),粒径越小,比表面积越大,则活性基团暴露越多,进而影响其理化性质。咖啡果皮可溶性膳食纤维中值粒径显著大于其余5 种样品,可能由于采用传统提取方式,所得样品粒径大小分布不均。由图1也可看出,咖啡果皮可溶性膳食纤维粒径分布范围较宽,均一性较差,大枣可溶性膳食纤维分布最均匀。
2.2 单糖组成
多糖具有降糖、降脂、抗氧化及免疫调节等生物活性,这些生物活性与单糖组成密不可分。果胶主要由半乳糖醛酸、半乳糖、鼠李糖、木糖及阿拉伯糖组成,纤维素主要由葡萄糖组成,半纤维素主要由木糖、葡萄糖、甘露糖和半乳糖组成。图2 为10 种单糖的标准曲线及咖啡果皮可溶性膳食纤维的单糖色谱图,由表2 可知,不同来源可溶性膳食纤维在单糖种类、含量方面均存在差异。其中咖啡果皮可溶性膳食纤维的单糖种类最多,检测出10 种单糖,半乳糖醛酸含量最高达1.04 g/100 g,表明水提法制备的咖啡果皮可溶性膳食纤维主要成分为果胶,还包含部分纤维素及半纤维素。燕麦、大枣和芹菜可溶性膳食纤维中半乳糖含量最高,占比高达87%以上,表明其主要组成为半纤维素及果胶物质。大豆和菊粉可溶性膳食纤维中含量最高的为葡萄糖, 分别为2.18 、2.12 g/100 g,且占比为66%,表示其纤维素含量较高。
2.3 结构分析
2.3.1 傅里叶变换红外光谱(FT-IR)分析 FT-IR是研究多糖类化合物官能团及化学键常用的方法之一。由图3 可知,6 种可溶性膳食纤维均具有典型的多糖吸收峰,除了某些特征波段的强度不同之外,6 种样品具有相似的光谱分布,样品在3365.4 cm–1附近显示宽而强烈的特征峰,是O-H的伸缩振动形成的吸收峰[21],另外5 种可溶性膳食纤维在3365.4 cm–1的吸收峰均强于咖啡果皮可溶性膳食纤维,表明其氢键含量较高。2933.3 cm–1处的弱峰是由亚甲基C-H 伸缩振动引起,代表半纤维素的典型结构。1681.1 cm–1处的峰可能是由于糖醛酸的C=O 伸缩振动产生,1411.8 cm–1处的峰可能与O-H 的伸缩振动和C-H 的弯曲振动有关[22]。此外,950~1200 cm–1范围内的吸收峰被认为是碳水化合物的特征区域[23],可以识别糖类的组成,在这一区域,6 种可溶性膳食纤维谱图表现出细微差异,与其单糖组成具有一定的差异结果一致。
2.3.2 扫描电镜(SEM)分析 SEM 是研究膳食纤维微观结构的最重要方法之一。由图4 可知,咖啡果皮可溶性膳食纤维为不规则块状结构、大小不一、表面有不规则褶皱和蜂窝状小孔。大豆可溶性膳食纤维具有较光滑的块状结构,表面有不规则褶皱及小孔使其表现出吸附作用。菊粉为光滑的块状结构、表面褶皱及小孔较少,表面积较小。大枣、燕麦、芹菜均为不规则块状结构、表面褶皱及小孔较多,但颗粒大小不同,与粒径分析结果一致。以上结果表明,不同来源可溶性膳食纤维具有不同形态结构。
2.3.3 X-射线衍射(XRD)分析 XRD 是通过X-射线在晶体中所产生的衍射现象反映样品的结晶特性及结晶度。其峰形可以表示晶体类型,衍射峰强度表示结晶度,结晶度对食品材料力学及热性能具有很大影响。由图5 可知,咖啡果皮、大豆、菊粉、大枣、燕麦和芹菜6 种样品的结晶度分别为38.8%、42.6%、45.5%、46.1%、49.5%、48.3%。6 种可溶性膳食纤维均在20°附近有显著的结晶衍射峰,被认为是典型纤维素Ⅰ型结构,咖啡果皮可溶性膳食纤维在此处的衍射强度显著低于其余5 种样品,在14.2°、29.5°、30.6°和32°等附近还存在一些较小的弥散衍射峰,表明其存在部分结晶[8],6 种样品的结晶度,以及在结晶区与非晶区的分布情况具有一定的差异。
2.3.4 热稳定性分析 热稳定性是评价物料应用于食品中的一个重要指标,可根据物质热变性过程中温度和能量的变化来研究其结构稳定性和构象的变化。由图6 可知,6 种不同来源可溶性膳食纤维样品的热流曲线均表现为吸热峰,吸热峰的峰值代表了变性温度,峰值的差异可能与氢键的类型及数量有关,峰值越高,则需要更多的能量来破坏氢键[24]。菊粉可溶性膳食纤维峰值在110℃左右,其余5 种样品的吸热峰的峰值均在120℃左右。6 种样品的焓变值(ΔH)均在160.41~329.93 J/g之间,焓变值越大,表示其热稳定性越好,热流强度越大则表示其热稳定性越流强度低于其他几种可溶性膳食纤维样品,说明咖啡可溶性膳食纤维样品的热稳定性优于其他几种膳食纤维样品。
2.4 理化性质分析
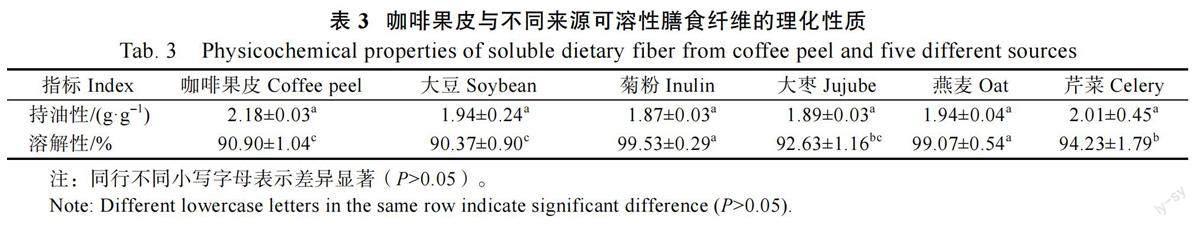
溶解性、持油性是评价可溶性膳食纤维的重要指标。持油性有利于食品在加工过程中保留油脂,在肠道中发挥调节代谢作用。表3 为6 种不同来源可溶性膳食纤维的持油性及溶解性,6 种样品的持油性无显著差异,但咖啡果皮(2.18 g/g)与芹菜可溶性膳食纤维(2.01 g/g)略高于其他4 种样品,可能是由于其表面褶皱较多,表面积较大。溶解性的高低与粒径大小及结构密切相关,菊粉可溶性膳食纤维的溶解性最高,为99.53%±0.29%,其次为燕麦样品(99.07%±0.54%),而咖啡果皮与大豆可溶性膳食纤维溶解性较小,这可能与其粒径分布有关。咖啡果皮可溶性膳食纤维粒径分布较广,均一性较差,因此其溶解度相对较低。
2.5 功能特性分析
2.5.1 胆固醇吸附能力 大量研究表明膳食纤维能降低血清总胆固醇的含量保护心血管[25]。由图7A 可知,6 种不同来源的可溶性膳食纤维对胆固醇的吸附能力存在显著差异。大枣可溶性膳食纤维的胆固醇吸附量最高为33.65 mg/g,菊粉的胆固醇吸附量最低为1.82 mg/g;刘晓贺等[26]对比研究豌豆与菊粉物化特性差异,其中菊粉对胆固醇吸附量为2.24 mg/g。咖啡果皮可溶性膳食纤维的胆固醇吸附量为22.43 mg/g,高于大豆及菊粉,但低于大枣、芹菜、燕麦可溶性膳食纤维,具有作为优质膳食纤维来源的潜质。
2.5.2 胆酸钠吸附能力 胆汁酸由肝脏内合成并在胆囊中储存,在食物刺激下从胆囊中进入小肠,参与肝肠循环起到调节胆固醇代谢的作用。研究表明胆酸钠的吸附能力随其用量的增加而增强,胆酸钠浓度越高,膳食纤维的吸附能力越强。由图7B可知,在pH 为7.0 时,6 种不同来源可溶性膳食纤维对胆酸钠的吸附能力具有显著差异,大豆、大枣、燕麦、芹菜可溶性膳食纤维的吸附量高于咖啡果皮与菊粉,可能是由于胆酸钠浓度较低。
2.5.3 葡萄糖吸附能力 葡萄糖吸附能力也是膳食纤维的功能特性之一。可溶性膳食纤维可与肠液中的葡萄糖结合,导致餐后血糖水平下降,達到降血糖的目的。由图7C 可知,6 种样品的吸附量大小依次为:大豆(43.50 mg/g)>芹菜(33.85 mg/g)>咖啡果皮(27.40 mg/g)>菊粉(27.06 mg/g)>燕麦(24.10 mg/g)>大枣(23.80 mg/g),其吸附量存在差异,可能也与葡萄糖浓度有关。郭增旺等[27]研究了不同粒度大豆皮的理化及功能特性,结果表明样品对葡萄糖的吸附量可随葡萄糖浓度的升高而升高,浓度越高,与样品的网状结构接触几率越大,对葡萄糖的束缚力就会增强。
2.5.4 亚硝酸盐吸附能力 在胃酸环境中,亚硝酸盐与仲胺、叔胺、酰胺反应生成强致癌作用的亚硝胺化合物。研究表明,亚硝酸盐在pH 为2.0条件下的吸附量显著大于pH 为7.0 时的吸附量。由图7D 可知,6 种不同来源的可溶性膳食纤维在胃酸环境中吸附亚硝酸盐的能力显著高于小肠环境。在pH 为2.0 时,咖啡果皮可溶性膳食纤维的吸附量为7.93 mg/g,显著高于其他5 种样品。在pH 为7.0 时,咖啡果皮、菊粉、大枣、燕麦可溶性膳食纤维的亚硝酸盐吸附量高于芹菜与大豆样品。以上结果表明,pH 对样品的亚硝酸盐吸附量影响较大,不同来源可溶性膳食纤维在相同pH条件下,吸附量也存在显著差异。
3 讨论
本研究对比了6 种不同来源可溶性膳食纤维理化性质、结构及功能特性方面的差异。膳食纤维被归类为从植物细胞壁中提取的多种不可消化营养素,被称为生物体的第7 种重要营养素,在机体健康方面扮演着重要的角色,其中可溶性膳食纤维具有更高的生理功能。可溶性膳食纤维的来源不同其结构、理化性质及功能特性存在显著差异。罗白玲等[18]研究了超微粉碎对咖啡果皮膳食纤维结构及性质的影响,其中超微粉碎可以有效改善样品的功能性质,但本研究中粒径大小与功能特性无显著关联,可能是由于样品的某些特性不仅与粒径大小有关,还与样品的来源、提取方法、加工参数以及实验条件等因素有关。本研究中6 种可溶性膳食纤维在结构方面均具有多糖的典型特征,其扫描电镜结果与栗俊广等[28]在研究鹰嘴豆、麦麸、大豆、葡萄膳食纤维性质差异的研究中得到的结果类似。本研究中不同来源的可溶性膳食纤维在功能特性方面也表现出显著差异,咖啡果皮可溶性膳食纤维在亚硝酸盐吸附作用中表现优异,与GAN 等[29]以葡萄柚皮为原料,探究微波辅助提取法对其结构及功能特性的影响研究中的结果类似,亚硝酸盐的吸附能力在pH 2.0 时显著强于pH 7.0。这可能是因为胃酸环境中存在的酚酸如阿魏酸,有利于增强亚硝酸盐的清除能力,使样品的吸附能力更强。其余几种吸附能力可能也与相关实验参数有关,如钟希琼等[30]在研究麦麸、米糠、豆渣、甘薯、大薯、葛根、香芋和马铃薯8 种膳食纤维的胆酸钠吸附能力时, 胆酸钠浓度为2.0 mg/mL 与3.0 mg/mL 时膳食纤维的吸附量存在显著差异。综上所述,咖啡果皮可溶性膳食纤维作为咖啡副产物,具有较高的利用价值,为后续咖啡副产物的精深加工提供了研究方向。
4 结论
本研究对比了咖啡果皮可溶性膳食纤维与大豆、菊粉、大枣、燕麦、芹菜5 种市售可溶性膳食纤维的粒径、单糖组成、微观结构、理化性质及功能特性。明晰了传统方法提取的咖啡果皮可溶性膳食纤维与常见豆类、果蔬膳食纤维的差异,为咖啡果皮可溶性膳食纤维的改性、精深加工提供了方向。研究得出:(1)6 种可溶性膳食纤维的粒径分布不同,其中咖啡果皮可溶性膳食纤维分布较广,均一性比其余5 种样品差,对样品的溶解性具有一定影响;咖啡果皮可溶性膳食纤维共检出10 种单糖,含量最多的为半乳糖醛酸,种类不同含量不同,表示其纤维素、半纤维素、果胶物质含量不同。(2)红外光谱结果表示具有典型的多糖吸收峰,在特征波段的强度与指纹区域略有差异,与单糖组成结果一致;扫描电镜结果表明不同来源可溶性膳食纤维,形态结构略有差异。6 种样品均为不均匀块状结构,表面有小孔及褶皱,咖啡果皮可溶性膳食纤维小孔更为清晰,表面褶皱较多,大豆可溶性膳食纤维与菊粉表面则较为光滑,褶皱小孔较少;X-射线衍射结果表明6 种样品均存在纤维素Ⅰ型结构,且咖啡果皮结晶度最小为38.8%,并且其热稳定性优于其他5种样品。(3)不同样品间持油性无显著差异,菊粉的溶解性最高为99.53%,咖啡果皮持油性最低为90.9%。(4)咖啡果皮可溶性膳食纤维的亚硝酸盐吸附能力显著优于其他5 种样品,吸附量为7.93 mg/g。总体来说,咖啡果皮可溶性膳食纤维较其余5 种膳食纤维具有更丰富的单糖组成,较高的亚硝酸盐吸附能力以及热稳定性,本研究可为咖啡果皮资源的开发利用提供理论基础,为咖啡果皮可溶性膳食纤维的高值化利用提供理论依据和技术支撑。
