基于SLAF-seq技术的苹果SNP位点开发及遗传多样性分析
2023-05-23刘万达杨光焦奎宝王昆王天鹤王禹吴立仁
刘万达 杨光 焦奎宝 王昆 王天鹤 王禹 吴立仁


摘要:为了解黑龙江苹果种质资源的遗传关系,利用特异性位点扩增片段测序技术(SLAF-seq)对在黑龙江地区收集的31份苹果种质资源进行SNP 位点开发和遗传多样性分析。最终获得了107.52 Mb读长数据,不同材料的SNP标记数目在 309 012~540 030 间,样品测序质量值平均Q30为93.78%,平均GC含量为40.90%。共开发获得 1 072 115 个SLAF标签,其中,多态性SLAF标签有275 389 个。通过序列分析,共得群体SNP位点121 352 个,基于开发SNP分子标记,结果表明,31 份苹果种质资源分为3 个亚群,特异性的苹果SNP位点开发和研究可为苹果种质资源鉴定和遗传多样性分析提供理论基础。
关键词:苹果;SLAF-seq技术;SNP;遗传多样性分析
中图分类号:S661.102 文献标志码:A
文章编号:1002-1302(2023)08-0061-06
基金项目:农业农村部园艺作物种质资源利用重点实验室开放基金(编号:NYZS201906);黑龙江省自然科学基金联合引导项目(编号:LH2022C097)。
作者简介:刘万达(1982—),男,黑龙江齐齐哈尔人,硕士,副研究员,主要从事寒地苹果育种及栽培技术研究。E-mail:haaslwd@163.com。
苹果(Malus pumila MILL.)是我国主要栽培果树树种之一,分布广、品种多,具有很高的经济价值,与柑橘、葡萄、香蕉并称为世界四大水果[1]。黑龙江省中小型苹果由于干物质含量高,口味纯正,品质佳,深受广大消费者喜爱与认可。这些苹果种质资源中较大部分是由栽培大苹果品种与野生资源或地方品种杂交获得。两者基因的整合,获得的苹果资源既具有果实商品性又能够适应黑龙江省的环境。但经过几代杂交,近年黑龙江省培育的苹果新品种存在果实品质提高幅度不大、抗寒能力下降明显、适宜栽植区域小等问题,育种工作进展缓慢。地方品种是非常重要的种质资源,因此,研究分析其种质资源群体结构及遗传多样性对资源利用、种质鉴定及现代遗传育种具有十分重要的意义。
特异性位点扩增片段测序(SLAF-seq)是一种基于高通量测序技术发展而来的简化基因组测序技术,具有成本低、通量高、深度高、准确性高等突出优势,已被广泛用于作物遗传图谱构建、遗传多样性分析、进化关系分析和基因定位[2-4]。当前,基于SLAF-seq技术在紫苏、甘薯和金花茶[5-7]等作物上已开发大量的SNP标记及遗传进化分析,但在黑龙江省苹果种质上进行遗传多样性分析及大量SNP分子标记开发等尚未见报道。本研究利用SLAF-seq技术对来自黑龙江省的31份苹果种质资源进行遗传进化分析,探讨黑龙江省苹果种质资源的遗传背景及亲缘关系,为黑龙江省苹果品种的分类及鉴定奠定基础,为苹果种质资源的收集保存、开发利用及杂交组合选配提供科学的参考依据。
1 材料与方法
1.1 材料
试验于2021年6月从黑龙江省不同地区采集31份苹果种质资源嫩叶置于变色硅胶中保存备用,样品详细信息见表1。
1.2 DNA的提取和检测
供试苹果样本基因组DNA提取采用CTAB法,利用分光光度计检测DNA的纯度和浓度,利用1%琼脂糖凝胶电泳检测DNA完整性。
1.3 酶切预测
根据苹果基因组大小和GC含量等信息,本研究选用苹果基因组作为参考基因组(https://iris.angers.inra.fr/gddh13/the-apple-genome-downloads.html)进行酶切预测。组装获得的基因组709.56 Mb,GC含量为38.03%。最终确定选择 RsaⅠ+HaeⅢ为限制性内切酶切组合,最后选取目的片段回收,为确保酶切实验准确性,本研究选取粳稻品种日本晴为对照进行测序。利用Illumina平台对质量检测合格后的文库进行高通量测序,得到个体序列,最后对测序质量值 Q30和GC 含量进行评估和分析以保证测序质量。
1.4 SLAF 标签的获得及SNP 标记的开发
根据序列的相似性对31份苹果种质资源进行聚类分析,一个 SLAF 标签如果在不同苹果种质间的序列有差异,即为多态性SLAF 标签。依据 SLAF 标签开发 SNP 位点,利用BWA[8]将测序结果和参考基因组序列比对,利用GATK[9]和SAM tools[10]对SNP标记进行开发,最终选择2种方法获得的交集作为最可信的SNP标记。根据缺失率<20%、次要基因型频率(MAF)>5%的标准对所有的SNP位点进行过滤筛选。
1.5 遗传多样性分析
利用Admixture软件[11]对群体进行群体结构分析,利用MEGA5.0软件[12],通过Neighbor-joining[13]算法,对群体进行进化树和亲缘关系分析,利用Eigensoft软件[14]进行主成分分析。
2 结果与分析
2.1 酶切方案与建库评估
利用酶切预测软件分析,选择HaeⅢ+RsaⅠ为限制性内切酶,酶切片段大小在364~464 bp的序列定义为SLAF标签,共预测到123 512个SLAF标签。利用SOAP软件[15],将水稻日本晴的读长与参考基因组进行比对,结果(表2)表明,双端比对效率为94.72%,测序数据酶切比例为83.40%,说明比对效率基本正常。根据获得的SLAF标签的实际长度,绘制插入片段长度分布图,结果(图1)表明,读长插入片段在预期范圍(364~464 bp)之内,表明SLAF建库正常,可用于下步试验。
由测序碱基分布情况(图2)可知,碱基A与T、C与G分布较一致,符合碱基配对的原则,提示测序未出现错误,因此获得SLAF标签可用于SNP标记的开发和遗传多样性分析。
2.2 簡化基因组序列的产出和质量评估
对31份苹果种质的测序数据统计结果,由图3可知,共计获得58.21个百万读长序列(Mreads)数据,各样品所获得的读长数为823 725~3 077 993,其中,来源黑龙江省哈尔滨市的品系“1962”样品所获得读长数最大,为3 077 993个读长,来源于黑龙江省鸡西市的地方品种“一窜铃”样品所获读长数最小,为823 725个读长(图3-A)。测序质量值Q30为92.93%~94.67%,测序平均Q30为93.50%,其中,来源于黑龙江的地方品种“花红”样品Q30测序值最大,为94.67%,来源于内蒙古自治区的育成品种“塞外红”样品的Q30测序值最小,为92.93%。所有样品Q30值均在90%以上,说明测序数据合格。测序获得GC比例为40.09%~42.14%,其中,来源于黑龙江哈尔滨市的品系“245”样品的GC比例最大,为42.14%,来源于黑龙江绥棱的地方品种“绥棱黄果”样品的GC比例最小,为40.09%,平均GC含量为40.80%,GC比例普遍不高,说明达到测序要求(图3-B)。
2.3 SLAF标签和SNP分子标记的开发
通过序列分析,共开发获得了136 282个SLAF标签,其中多态性SLAF标签109 966个。由图4可知,样品的平均测序深度为11.29%(图4-A),从多态性SLAF标签中开发获得了2 039 575个SNP位点(图4-B),对所有的SNP根据次要基因型频率(MAF)>5%,缺失率<20%过滤,筛选高一致性的群体SNP。统计多态性SLAF标签和SNP标记在苹果不同染色体上的个数,根据数据绘制染色体分布图,由图5可知开发的多态性SLAF标签和SNP位点在苹果基因组17 条染色体上的分布情况,且在每条染色体上有比较均匀的分布,说明测序结果正常,可进行下一步分析。
2.4 群体遗传结构分析
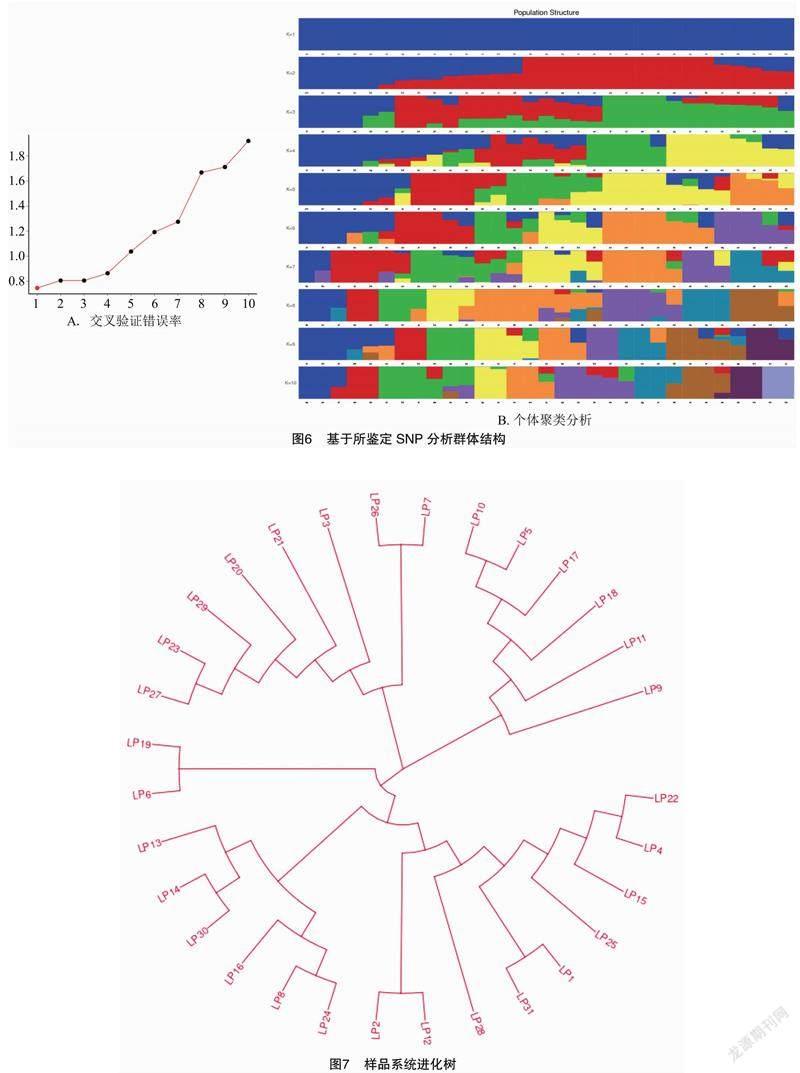
基于已开发的苹果SNP位点,利用Admixture软件分析了研究材料的群体结构。根据交叉验证错误率的谷值确定最优分群数。由聚类分析结果(图6)可知,K值为1时交叉验证错误率最低,可见31份苹果资源间遗传结构不明显,说明不同来源的个体来源于同一祖先。
基于已开发的苹果SNP位点,利用MEGA5软件绘制31份苹果种质的遗传关系聚类图,由图7可知,31份苹果资源可划分出3个差异性分支,第一个分支由17份苹果资源组成,其中9份育成品种,4份地方品种,2份品系,2份其他。第二个分支由8份苹果资源组成,其中2份育成品种,4份地方品种,1份品系,1份其他。第三个分支由6份苹果资源组成,其中4份育成品种,2份品系。说明种质间存在较大变异,具有丰富的遗传多样性。聚类结果表明黑龙江省苹果资源没有明显的区域特征。
基于已开发的苹果SNP位点,利用Eigensoft软件,对31个苹果种质进行主成分分析(PCA),得到样品的聚类情况。由图8可知,31份苹果种质聚为三维(PCA1、PCA2、PCA3),进一步提示黑龙江苹果31份种质分化为3个亚群,黑龙江省苹果资源品种之间存在遗传多样性。
3 讨论与结论
苹果是我国主要栽培果树树种之一,其面积和产量均居世界首位。目前,针对苹果开展遗传进化分析研究,主要集中在生理生化水平和表型鉴定等方面。SLAF-seq技术具有快速、低成本、高通量开发SNP标记的优势[16],国内外研究者利用该技术已经在广金钱草、山西省地方梨、白皮松等作物上成功应用[17-19],姜涛等运用SLAF-seq技术对39份连翘种质资源进行了SNP标记的开发和遗传多样性分析,共获得262 297个多态性SLAF标签,开发了1 809 741个SNP标记[20]。陶红霞等运用SLAF标记,获得125 497个多态性SLAF标签,构建了苹果遗传连锁图谱[21]。李敏等通过SLAF-seq技术测序,获得了99 526个多态性SLAF标签,并开发了9 488个高质量的SNP标记[22]。
植物的生长环境对物种的种群结构和遗传分化会产生直接的影响,通过苹果样品的系统进化树发现,黑龙江苹果种质遗传多样性较为丰富,育成品种中“大秋”和“秋露”亲缘关系比较近,地方品种中“冻果”和“山丁子”亲缘关系较近,而地方品种“红铃铛”和育成品种“七月鲜”亲缘关系较近,上述品种之间亲缘关系较近,可能是杂交育种产生的。本研究利用特异性位点扩增片段测序技术,通过对31份苹果种质资源进行SNP 位点开发和遗传多样性分析,共得到 107.52 Mb Clean Reads 数据,每个样品的 SNP 标记数目介于 309 012~540 030 之间,样本平均测序质量值 Q30 为93.78%,样品平均 GC 含量为 40.90%。共得到 1 072 115 个 SLAF 标签,其中275 389 个多态性 SLAF 标签。通过序列分析,获得 121 352 个有效的单核苷酸多态性标记(SNP),利用开发的SNP分子标记,将31份苹果种质资源分为3个亚群。本研究平均测序深度较高,获得的SNP位点数量多,为今后结合苹果果实性状、叶片性状等数量性状进行苹果属资源的评价利用,如亲本选择、关联定位分析、杂种优势利用及进化[23]等研究提供了重要的分子标记辅助育种标记,为苹果种质资源保护和品种创制等研究提供了有力证据。
参考文献:
[1]宋春晖,陈晓菲,王枚阁,等. 基于SLAF-seq技术鉴定苹果砧木耐涝候选基因[J]. 中国农业科学,2021,54(18):3932-3944.
[2]Dong Z M,Chen L,Li Z,et al. Identification and molecular mapping of the semi-dwarf locus (sdf-1) in soybean by SLAF-seq method[J]. Euphytica,2020,216(6):103.
[3]Wei Q Z,Wang W H,Hu T H,et al. Construction of a SNP-based genetic map using SLAF-seq and QTL analysis of morphological traits in eggplant[J]. Frontiers in Genetics,2020,11:178.
[4]Zhang S Z,Hu X H,Miao H R,et al. QTL identification for seed weight and size based on a high-density SLAF-seq genetic map in peanut (Arachis hypogaea L.)[J]. BMC Plant Biology,2019,19(1):537.
[5]姜 涛,刘灵娣,田 伟,等. 紫苏SNP分子标记开发及遗传多样性分析[J]. 分子植物育种,2021,19(4):1243-1249.
[6]苏文瑾,赵 宁,雷 剑,等. 基于SLAF-seq技术的甘薯SNP位点开发[J]. 中国农业科学,2016,49(1):27-47.
[7]刘 凯,李开祥,韦晓娟,等. 基于SLAF-seq技术的金花茶SNP标记开发及遗传分析[J]. 经济林研究,2019,37(3):79-83.
[8]Li H,Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform[J]. Bioinformatics,2009,25(14):1754-1760.
[9]McKenna A,Hanna M,Banks E,et al. The Genome Analysis Toolkit:a MapReduce framework for analyzing next-generation DNA sequencing data[J]. Genome Research,2010,20(9):1297-1303.
[10]Li H,Handsaker B,Wysoker A,et al. The sequence alignment/map format and SAMtools[J]. Bioinformatics,2009,25(16):2078-2079.
[11]Alexander D H,Novembre J,Lange K. Fast model-based estimation of ancestry in unrelated individuals[J]. Genome Research,2009,19(9):1655-1664.
[12]Tamura K,Peterson D,Peterson N,et al. MEGA5:molecular evolutionary genetics analysis using maximum likelihood,evolutionary distance,and maximum parsimony methods[J]. Molecular Biology and Evolution,2011,28(10):2731-2739.
[13]Saitou N,Nei M. The neighbor-joining method:a new method for reconstructing phylogenetic trees[J]. Molecular Biology and Evolution,1987,4(4):406-25.
[14]de Hoon M J L,Imoto S,Nolan J,et al. Open source clustering software[J]. Bioinformatics,2004,20(9):1453-1454.
[15]Li R Q,Yu C,Li Y R,et al. SOAP2:an improved ultrafast tool for short read alignment[J]. Bioinformatics,2009,25(15):1966-1967.
[16]段義忠,王建武,杜忠毓,等. 基于SLAF-seq简化基因组技术的沙冬青SNP位点开发及遗传分析[J]. 植物研究,2018,38(1):141-147.
[17]唐晓敏,张春荣,周良云,等. 基于SLAF-Seq技术的广金钱草SNP位点开发及遗传分析[J]. 分子植物育种,2020,18(18):6101-6107.
[18]白牡丹,郝国伟,张晓伟,等. 基于SLAF-seq技术的山西省地方梨品种的SNP分析[J]. 西北农业学报,2020,29(7):1020-1027.
[19]田 倩,刘双委,钮世辉,等. 基于SLAF-seq技术的白皮松SNP分子标记开发[J]. 北京林业大学学报,2021,43(8):1-8. [HJ2mm]
[20]姜 涛,温春秀,田 伟,等. 基于SLAF-seq技术连翘SNP分子标记开发及遗传多样性分析[J]. 分子植物育种,2021,19(16):5405-5413.
[21]陶红霞. 基于SLAF标记的苹果遗传连锁图谱构建[D]. 杨凌:西北农林科技大学,2015.
[22]李 敏,郭 聪,王 莹,等. 基于SLAF-seq技术的乔木柳SNP位点开发[J]. 西南农业学报,2018,31(5):891-895.
[23]李晓颖,郑少泉,徐红霞,等. 基于SLAF-seq技术枇杷SNP位点开发[J]. 分子植物育种,2021,19(15):5038-5045.
