基于化学计量方法甘松药材不同采收期挥发油及指纹图谱差异性评价
2023-05-21王继强王二欢杨祎辰马存德刘腾恬
王继强,王二欢,杨祎辰,马存德,刘腾恬,安 莉,李 婷,王 慧,刘 峰*
(1.陕西国际商贸学院 中药研究院,陕西 咸阳 712046;2.陕西步长制药有限公司,陕西 西安 710075)
甘松来源为败酱科甘松属植物甘松NardostachysjatamansiDC.的干燥根及根茎[1],别名甘松香,性味辛,甘,温,归脾胃,有理气止痛,开郁醒脾的功效,为我国二级保护藏药。对其记载最早见于《本草拾遗》[2],《海南本草》和《开宝本草》等也有记载。甘松多分布于喜马拉雅山脉南北两侧,主产于我国四川、甘肃、青海等地以及印度北部、尼泊尔[3]。品种划分中将其分为甘松、匙叶甘松Nardostachysjatamansi(D.Don) DC、大花甘松NardostachysgrandifloraDC,有学者认为甘松、匙叶甘松、大花甘松之间为性状过渡[4],均为同一种植物。
甘松有效成分研究中,多集中在挥发油成分方面[5]及非挥发性成分的环烯醚萜类、单帖、倍半萜、二萜、三萜、黄酮类等方面,其中萜类化合物为主要活性成分[6]。冯海生等[7]研究甘松不同来源不同生长发育阶段成分的动态变化,耿晓萍等[8]分析甘松不同部位挥发油化学成分对比研究及甘松新酮的提取工艺优化,表明不同阶段成分含量存在差异。刘国林等[9]通过测定黄酮及甘松新酮等物质评价海拔高度对甘松有效成分含量的相关性分析,结果表明总黄酮与甘松新酮与海拔高度具有一定相关性;伍杰等[10]研究分析不同产地甘松挥发油、甘松新酮、绿原酸、灰分、微量元素等指标的检测,表明其质量差异与生长环境密切相关。张宇霞等[11]依托高原生物研究所,论述不同采收期、不同产地甘松挥发油及浸出物存在较大的差异;买吾兰江·买提努尔等[12]开发甘松指纹图谱及甘松新酮色谱测定技术,杨祎辰等[13]采用Box-Benhnken响应面法提取优化甘松挥发油的提取方法。李莹[14]采用色谱技术评价甘松药效物质基础及质量标准的初步研究;张毅[15]采用现代技术评价化学成分的抗肿瘤活性化合物的合成及构效关系;李艳忙等[16]对其研究结果进行论述。
近年来随着研究深入,甘松有效成分测定也逐步标准化,2015版及后续《中华人民共和国药典》增加有效成分甘松新酮含量测定,尤其随着近年来产区政策影响,甘松药材肆意采挖,产量逐渐下降,各地区药材质量差异较大,难以把控品质。尤其在市场交易中,甘松多为全草入药,鲜有全根,为此,本研究以甘松栽培原材为样品,通过对不同部位不同采收期挥发油的测定及建立有效成分的指纹图谱全方位评价,探讨其动态变化过程及市场流通的合理性,对建立甘松药材种植技术质量评价体系提供一定的依据,诠释人工种植以及不同部位入药的可行性。
1 材料与方法
1.1 仪器与试剂
岛津2030Plus液相色谱仪(日本岛津);TE124S分析天平(德国Sartorius公司);DHG-9050B智能型电热恒温鼓风干燥箱(上海琅玕实验设备有限公司);KQ-800KDE高功率数控超声波清洗器(昆山市超声仪器有限公司);TQ-500高速多功能粉碎机(上海市天祺盛世科技有限公司);98-I-BN电热套(天津市泰斯仪器有限公司);挥发油提取装置(上海垒固仪器有限公司);乙腈、甲醇、异丙醇(色谱纯,德国MERCK公司);甲醇、无水乙醇、磷酸(分析纯AR,国药集团化学试剂有限公司);甘松新酮(AF21021603)、绿原酸(AF21012553)、蒙花苷(批号:AZ21091601)、隐丹参酮(批号:AF20092606),均来自成都埃法生物科技有限公司。
1.2 试验材料
于2022年3月-2022年10月,在步长制药甘肃药源基地采收甘松不同部位(根及根茎、茎叶、全草)样品,阴干后保留备用,采用仿野生栽培技术,引种驯化并采用分株繁殖方式种植完成生长周期,经陕西步长制药有限公司主任药师马存德鉴定为败酱科甘松属植物甘松NardostachysjatamansiDC.,详见表1。
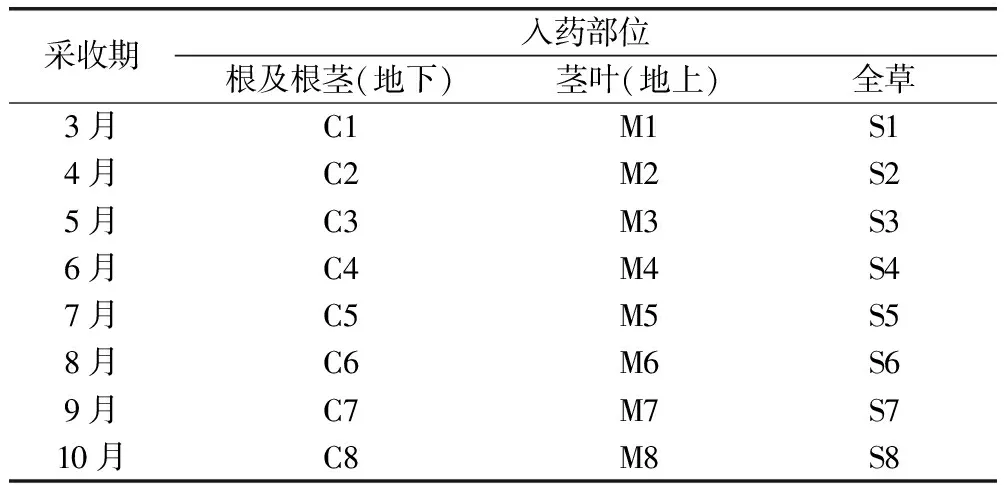
表1 样品批次编号
2 试验方法与结果
2.1 挥发油提取测定
试验样品阴干后打粉过3号筛,参考2020版《中华人民共和国药典》四部通则2204甲法,采用水蒸气蒸馏法提取挥发油,粒径527μm,料液比1∶10(g∶mL),提取6h,经无水硫酸钠干燥后,挥发油颜色为浅绿色至淡黄色油状,含有特殊香味。
2.2 色谱条件
色谱柱:Diamonsil Plus 5μm C18-A,250mm×4.6mm;岛津2030 Plus高效液相色谱仪;流动相:乙腈(A)-0.2%磷酸溶液(B);柱温:30℃;流速:1.0mL/min;进样量:10μL;波长275nm,洗脱条件见表2。

表2 梯度洗脱条件
2.3 对照品溶液制备
精密称取甘松新酮对照品适量,加甲醇制成每1mL中0.50mg溶液;精密称取绿原酸、蒙花苷、隐丹参酮对照品适量,加甲醇分别制成每1mL中含0.25mg的溶液,于4℃保存备用。
2.4 样品溶液的测定
精密称取甘松不同采收期药材粉末(过3号筛)1.00g,置于三角瓶中,以甲醇为溶剂,料液比1∶10,室温超声(800W,60kHz)提取60min,取上清液适量按照“2.2”方法,进行色谱测定,得甘松样品期不同部位指纹图谱及混合对照品图谱,见图1、图2。
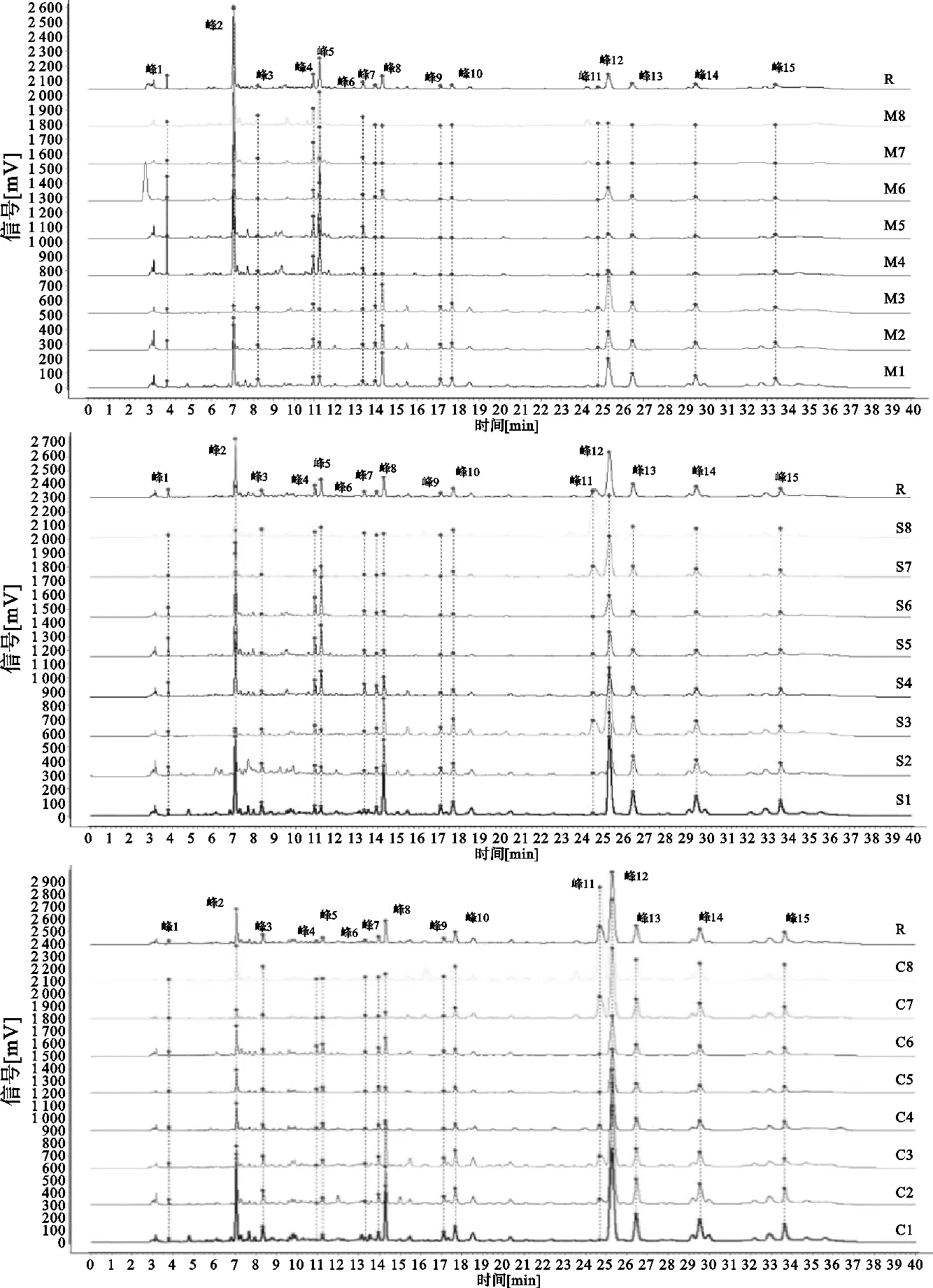
图1 甘松不同采收期不同样品指纹图谱
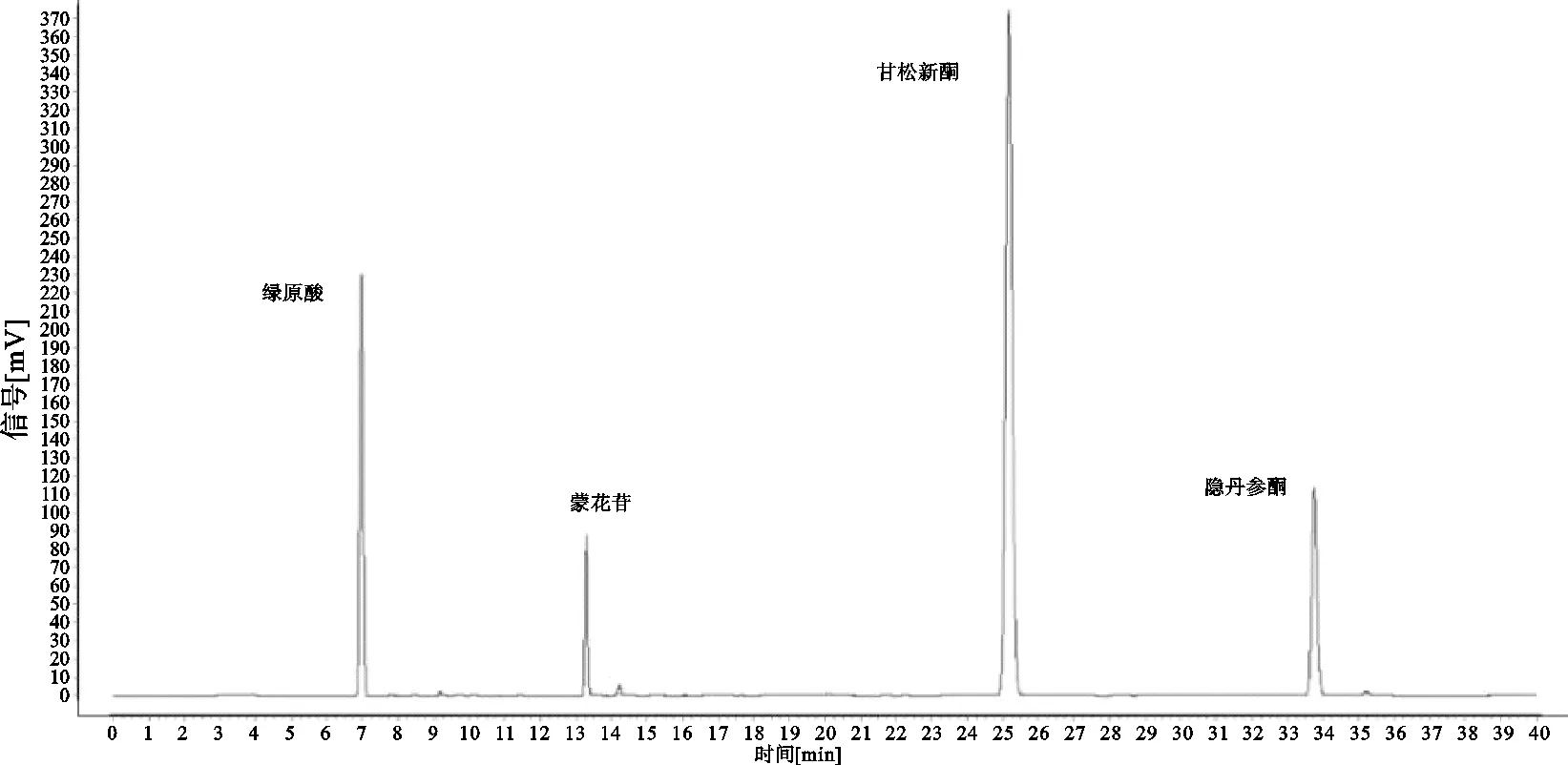
图2 甘松对照品图谱
2.5 方法学考察
2.5.1 精密度试验 精密吸取甘松溶液,在“2.2”色谱条件下,连续进样6次,以12号峰甘松新酮为参照峰,计算相对保留时间以及相对峰面积RSD值,结果表明各批次15个共有峰相对保留时间RSD为0.03%~0.46%,相对峰面积RSD为0.17%~1.45%。
2.5.2 重复性试验 按照“2.4”下平行称取甘松样品6次制成供试品溶液进行色谱测定,以12号峰甘松新酮作为参照峰,计算相对保留时间及相对峰面积的RSD值,结果表明各批次15个共有峰的保留时间RSD为0.13%~1.04%,相对峰面积RSD为0.22%~1.28%。
2.5.3 稳定性试验 取甘松供试品溶液,在“2.2”项色谱条件下,分别在0h、4h、8h、16h、20h、24h、48h进样,以甘松新酮为参照峰,计算相对保留时间及相对峰面积的RSD值,结果表明各批次15个共有峰的保留时间RSD为0.10%~1.28%,相对峰面积RSD为0.18%~2.02%。
2.5.4 线性考察 用甲醇稀释甘松对照品溶液甘松新酮,分别为原浓度的1、1/2、1/4、1/8、1/16、1/32倍,摇匀,经0.22μm滤膜过滤。在“2.4”项条件下,以对照品含量(mg/L)为横坐标X,测得的峰面积积分值为纵坐标Y,分别建立甘松新酮及绿原酸回归方程:Y=317.86X+1.913 4,R2=0.999 9;Y=37.372X+36.92,R2=0.999 9。
2.5.5 线性考察 用甲醇稀释对照品溶液,分别为原浓度的1、1/2、1/4、1/8、1/16、1/32倍,摇匀,经0.45μm滤膜过滤,备用。在“2.2”项条件下,以对照品含量(mg/L)为横坐标X,测得峰面积积分值为纵坐标Y,建立甘松新酮回归方程:Y=317.86X+1.913 4,R2=0.999 9。
2.6 数据处理
将不同采收期甘松样品色谱图导入“中药指纹图谱相似度度软件评价系统(2012.130723)”进行指纹图谱相似度评价,采用SPSS 26.0软件进行显著性差异分析、聚类分析以及主成分分析。
2.7 化学计量学分析
2.7.1 不同采收期挥发油含量差异分析 对不同采收期不同部位甘松挥发油含量进行非参数检验,采用傅莱德曼双向按秩方差分析,在显著性水平0.05下做双侧检验,显著性水平为0.03,存在显著性差异,挥发油含量最高为8.5%,最低为1.1%,皆为9月份样品,3月份全草高于根及根茎,7月份茎叶高于全草,其余月份为根及根茎大于全草大于茎叶;采用中位数进行聚类分析平方欧式距离,将其分为两类,9月份为一类,其余月份为二类,见图3;除5月、9月茎叶外,其余采收期挥发油含量都符合药典规定。
图3 甘松挥发油差异分析及聚类分析
2.7.2 不同采收期甘松不同部位HPLC指纹图谱建立 采用“中药指纹图谱相似度度软件评价系统(2012.130723)”分别对甘松不同采收期不同部位指纹图谱进行共有峰确定以及相似度评价,采用平均数法,经峰匹配后生成对照图谱R,见图1,甘松不同采收期不同部位样品HPLC图谱相似度结果见表3。
结果表明甘松不同采收期不同部位24批次样品中,在15个共有峰下,第2号峰为绿原酸,第6号峰蒙花苷,第12号峰为甘松新酮,15号峰为隐丹参酮,相似度范围在0.500~0.993之间;其中茎叶的相似度为0.500~0.986,根及根茎的相似度为0.586~0.994;全草相似度为0.533~0.992;根及根茎的相似度高于全草、茎叶。共有峰面积介于34.89%~74.79%,其中根及根茎为47.96%~74.79%,显著高于全草与茎叶。

表3 相似度数据
2.7.3 聚类分析及主成分分析 以甘松不同采收期不同部位共有峰峰面积为基准,导入SPSS 26.0分析软件,对其进行综合评价,采用中位数联结,欧式平方距离为分类标准进行系统分类;聚类分析结果见图4,聚类分析将其1~15共有峰分为3类,其中,2号峰、12号峰单独分为第一类、第二类、其余共有峰为第三类,由此可知,其特征峰2号绿原酸与12号甘松新酮为甘松药材的主要成分具有合理性,与相似度评价具有一致性。
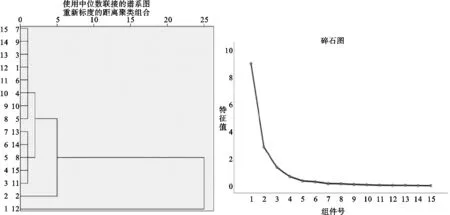
图4 聚类分析及碎石图
在聚类分析基础上,以各批次共有峰峰面积为基础进行主成分分析,其碎石图见图4,特征值及其累计方差贡献率见表4,以特征值>1为标准,共确定3种主成分,累计贡献率为87.794%,能够较好体现其成分特征;对主成分载荷值进行主成分计算的综合评价见表5,综合得分高低代表药材品质的差异,表明不同采收期对其影响较大;以3个主成分为基准得到主成分载荷图见图5,峰2(绿原酸)有较大的权重比,峰12(甘松新酮)落在第3主成分,峰15(隐丹参酮)落在第1主成分,表明在不同批次中成分相对稳定;从表5中可以看出,甘松不同采收期主成分综合得分评价中根及根茎优于全草优于茎叶,其中3月份样品中根及根茎的综合得分最高,其次为3月全草、9月根及根茎,由此可知,3月采收药材综合评价最优。
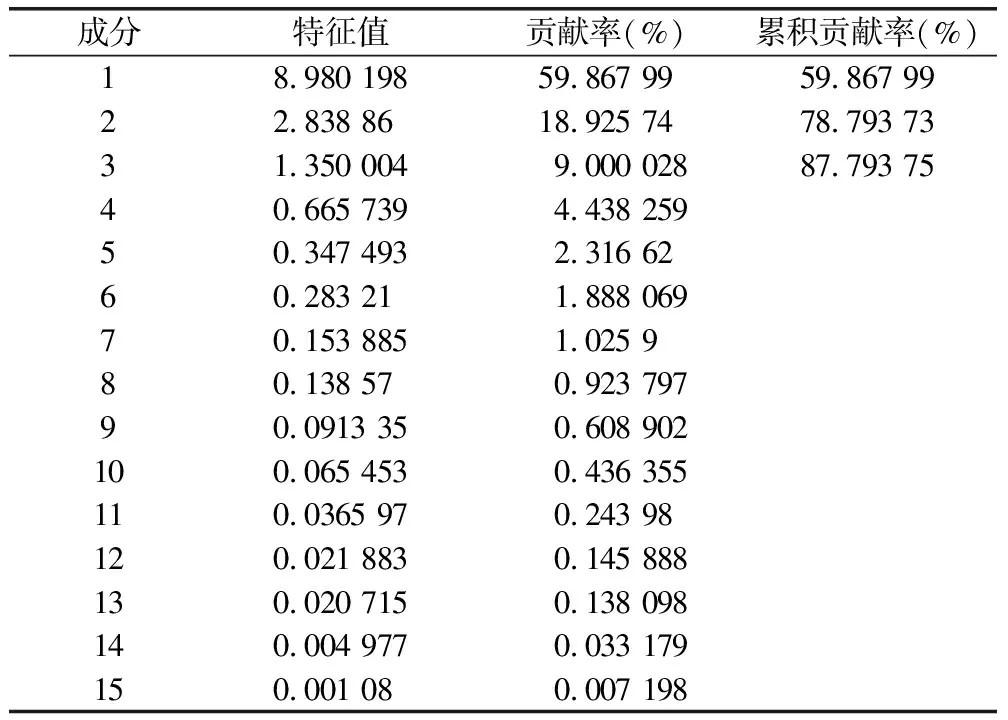
表4 主成分特征值及贡献率

表5 不同采收期主成分因子及综合评价
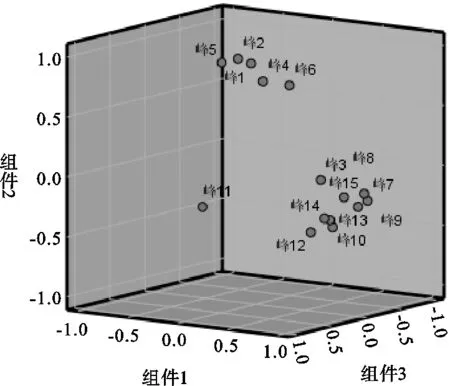
图5 主成分分析
3 讨论
本试验采用中药指纹图谱技术结合化学计量分析方法,在市场药材流通及产地分布、资源蕴藏量调研前提下,开展甘肃产地甘松栽培药材采收期试验研究,测定甘松挥发油含量。采用聚类分析、显著差异评价不同采收期挥发油动态变化规律;采用傅莱德曼双向按秩方差分析进行挥发油的显著性分析,在显著性水平0.05下做双侧检验,显著性水平为0.03,聚类分析中分为两类,其中9月份单独为一类。同时结合HPLC相似度评价及化学计量分析方法能够更好体现数据模型的合理性。
在对干松不同采收期不同部位指纹图谱评价过程中,根据研究目的,建立不同采收期HPLC指纹图谱,对其进行聚类分析、主成分分析,在极大程度上反映甘肃产区甘松样品品质的稳定性;由此可知,在不同采收期中,主成分种类具有一致性,相对含量随着季节不同有所变化,其中地下部分相对含量高于全草,高于茎叶;共有峰面积占比为47.96%~74.79%,主成分分析中3个主成分累计贡献率为87.793 75,主成分因子综合评价评价中根及根茎>全草>茎叶,可以客观反映药材品质的整体性与差异性。
不同采收期共有峰峰面积变化趋势与植物生长量、物质转移等具有相关性,不同部位之间挥发油变化差异有一致性,挥发油含量最高为9月份,有效成分含量主成分分析最佳为3月份,综合当地气候环境及采收成本问题以春季3月土地解冻后采收最佳。
样品采集方法中,以植物生长周期为基准,从出苗期到枯萎期,夏季绿原酸含量不断增加,初春及秋末甘松新酮含量较高且趋势明显,其中10月末由于环境原因成分出现分解现象,这一变化趋势与气候等自然因素有关外还有待进一步研究,次生代谢产物积累规律中,由于栽培技术不成熟,叶冠比、根的分支等与野生差异较大,尚未全面推广。市场中甘松药材多为全草入药,少有全根,本试验以甘肃产区栽培品为样品论述不同采收期不同部位入药的合理性及差异性,表明全草入药具有可行性,为其资源评价,综合开发利用,药材稳定性提供研究基础和方法,为优质甘松药材栽培提供可行的参考与依据。
