MiR-103a通过TAK1介导的NF-κB信号通路保护炎症诱导的心肌细胞损伤的研究
2023-05-18李璐斐金培印王磊
李璐斐,金培印,王磊
心血管疾病是目前引起人类疾病死亡第二多的疾病,包括心肌梗死、动脉粥样硬化、高血压、心力衰竭、心律失常以及高脂血症等,而这些心血管疾病通常伴随心肌炎症[1,2]。心肌细胞长期处于这种炎症环境下,可导致心肌细胞损伤,最终引起心肌细胞凋亡;心肌细胞凋亡不仅会直接影响正常的心脏功能,严重者还会导致患者死亡,而炎症诱导的心肌细胞凋亡主要受细胞内的基因调控[1,2]。因此,探究炎症环境下心肌细胞的凋亡机制对于预防和治疗心血管疾病具有重要意义。
微小RNA(miRNA)是一段长为22个核苷酸左右的非编码小RNA,它们虽然不能直接编码蛋白质,但可通过对与编码蛋白的基因序列3'端非编码区结合而影响其转录,进而参与调控细胞的生物学特征[3]。近年研究发现,miRNA通过对其靶基因的调控而参与恶性肿瘤[4]、心肌缺血再灌注损伤[5]以及脑缺血损伤[6]等多种疾病的发生发展。此外,大量研究发现,多种miRNA在急性心肌梗死患者外周血表达异常,包括miR-150[7]和miR-124-3p[8]。近期研究发现[9],miR-103a在败血症患者外周血中表达降低,并可通过抑制HMGB1的表达而抑制外周血单核细胞炎症。此外之前有研究表明[10],miRNA-103a通过抑制Atg5而抑制心肌细胞自噬和凋亡。然而,miR-103a的表达对炎症诱导的心肌细胞凋亡的影响还未见文献报道。本研究在体外通过肿瘤坏死因子(TNF-α)诱导的H9C2心肌细胞损伤建立炎症诱导的心肌细胞损伤体外模型,通过过表达miR-103a来研究miR-103a表达对TNF-α诱导的H9C2心肌细胞凋亡的影响,并探讨其调控分子机制,为开发靶向miR-103a治疗心血管疾病提高理论依据。
1 材料与方法
1.1 材料H9C2心肌细胞购买自武汉普诺赛生命科技有限公司;RIPA裂解液、Steady-Lumi™ Ⅱ萤火虫萤光素酶报告基因检测试剂盒、Annexin V-FITC细胞凋亡检测试剂盒、BCA蛋白浓度测定试剂盒、caspase 3、caspase 8及caspase 9活性检测试剂盒均购买自上海碧云天生物科技有限公司;PrimeScript™ RT reagent kit和qPCR Master Mix购买自宝日医生物技术(北京)有限公司;Lipofectamine 2000、DMEM高糖培养基、胎牛血清、miR-nc和miR-103a-mimic(miR-103a)购买自赛默飞世尔科技有限公司;PCR引物、si-nc和si-TAK1由生工生物工程(上海)股份有限公司官方设计合成;TAK1抗体、NF-κB抗体和p-NF-κB抗体、GAPDH抗体及HRP标记的羊抗鼠抗体均购买自艾博抗(上海)贸易有限公司。
1.2 实验方法
1.2.1 细胞培养与炎症诱导模型H9C2心肌细胞培养在添加10%胎牛血清的DMEM培养基中,培养条件为37℃+5%CO2。在H9C2心肌细胞培养基中添加20 μg/ml TNF-α后,正常培养24 h以建立炎症诱导心肌细胞损伤体外模型。
1.2.2 细胞转染运用Lipofectamine 2000转染将miR-nc、miR-103a、si-nc以及si-TAK1单独转入到H9C2心肌细胞中。转染8 h后,将细胞培养基更换新鲜培养基,48 h后通过检测miR-103a和转化生长因子β活化激酶1(TAK1)表达以验证转染效率。同时,转染48 h后参考1.2.1所述建立心肌细胞炎症诱导损伤模型。
1.2.3 实时荧光定量PCRH9C2细胞经处理后被收集,加入trol以提取细胞总RNA,然后使用PrimeScript™ RT reagent kit试剂盒合成cDNA,参考qPCR Master Mix试剂盒说明书制备20 μl RT-qPCR系统,并使用ABI 7500荧光定量PCR仪器检测miR-103a的相对表达量。miR-103a引物为:正向:5'- ACACTCCAGCTGGGAGCAGCATTGTACA GGG-3';反向:5'- TGGTGTCGTGGAGTCG-3';以U6作为内容,U6引物为:上游引物5'-CTCGCTTCGGCAGCACA-3';下游引物5'-AA CGCTTCACGAATTTGCG-3'。
1.2.4 Western BlotH9C2细胞经处理后被收集,加入RIPA裂解液以提取细胞总蛋白,经BCA蛋白浓度测定试剂盒测定总蛋白浓度,使用10%SDSPAGE蛋白分离胶分离50 μg总蛋白,再经转膜后,室温封闭2 h,4℃孵育一抗过夜,室温孵育二抗1 h后显影。以目的蛋白条带与GAPDH蛋白条带灰度值比值表示TAK1、NF-κB和p-NF-κB蛋白表达水平。
1.2.5 凋亡相关蛋白检测H9C2细胞经处理后被收集,加入RIPA裂解液裂解细胞,离心以收集细胞裂解液上清,最后运用caspase 3、8或9活性检测试剂盒检测细胞裂解液中caspase 3、8或9蛋白浓度,以caspase 3、8或9蛋白浓度与总蛋白浓度的比值表示caspase 3、8或9蛋白相对表达水平。
1.2.6 细胞活性与凋亡检测H9C2细胞经处理后被收集,参考CCK8试剂盒说明书检测H9C2细胞活性,运用Annexin V-FITC细胞凋亡检测试剂盒检测H9C2心肌细胞凋亡率。
1.2.7 双荧光素酶报告基因检测通过Target Scan网站(结合NCBI查找TAK1基因3'-UTR与miR-103a结合的部位,将这段3'-UTR的野生型(WT)和突变型(MUT)克隆到经Sac I和Sal I双酶切后的pmirGLO质粒上,将构建的重组质粒与miR-103a-mimic或者miR-nc共转到H9C2心肌细胞内,转染48 h后,收集细胞通过Dual-Luciferase系统(Promega,中国)检测荧光素酶荧光强度。
1.2.8 统计学分析采用SPSS 20.0统计学软件进行数据分析,计量资料采用均值±标准差表示,两组间差异由Student's t检验进行比较,多组间差异通过单因素方差分析进行比较。以P<0.05为差异具有统计学意义。
2 结果
2.1 TNF-α诱导对心肌细胞miR-103a和TAK1蛋白表达的影响经免疫印迹法检测H9C2细胞中TAK1蛋白表达,结果显示:与对照组相比,经TNF-α处理的TNF-α组H9C2心肌细胞中TAK1蛋白表达显著升高(图1)。经qRT-PCR法检测心肌细胞中miR-103a表达,结果显示:与对照组相比,经TNF-α处理的TNF-α组H9C2心肌细胞中miR-103a表达显著降低,表1,差异有统计学意义(P<0.05)。

图1 TNF-α处理后心肌细胞中TAK1蛋白的表达

表1 TNF-α处理后心肌细胞中miR-103a和TAK1蛋白表达
2.2 miR-103a过表达对TNF-α诱导的心肌细胞凋亡的影响运用CCK-8试剂盒检测H9C2心肌细胞的细胞活性,结果显示:与对照组相比,经TNF-α诱导的转染miR-nc的TNF-α+miR-nc组H9C2心肌细胞活性显著降低(P<0.05);且TNF-α+miR-103a组H9C2心肌细胞活性显著高于TNF-α+miR-nc组(P<0.05)。运用流式细胞仪检测心肌细胞的细胞凋亡率,结果显示:与对照组相比,经TNF-α诱导的转染miR-nc的TNF-α+miR-nc组H9C2心肌细胞凋亡率显著升高(P<0.05);且TNF-α+miR-103a组H9C2心肌细胞活性显著低于TNF-α+miR-nc组(P<0.05),表2。此外,与TNF-α+miR-nc组相比,TNF-α+miR-103a组中caspase 3、8和9蛋白在H9C2心肌细胞表达显著降低(P<0.05),表3。

表2 miR-103a过表达抑制TNF-α诱导的心肌细胞活性和凋亡

表3 miR-103a过表达抑制TNF-α诱导的心肌细胞凋亡相关蛋白活性
2.3 miR-103a靶向抑制心肌细胞TAK1蛋白表达在Target Scan 网站(www.targetscan.org)上可查询到miR-103a与TAK1蛋白存在互补序列(图2)。与miR-nc组相比,miR-103a组H9C2心肌细胞TAK1蛋白表达降低(P<0.05)(图3)。双荧光素酶基因报告细胞检测显示:与miR-nc组相比,miR-103a组WT处理的H9C2细胞荧光活性显著降低(P<0.05),而MUT处理的H9C2细胞荧光活性并未显著降低(P>0.05),表4。

表4 miR-103a对心肌细胞荧光素酶活性的影响

图2 miR-103a与TAK1基因互补序列
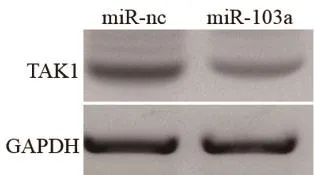
图3 miR-103a过表达抑制心肌细胞TAK1蛋白表达
2.4 TAK1敲低对TNF-α诱导的心肌细胞凋亡的影响运用CCK-8试剂盒检测H9C2心肌细胞的细胞活性,结果显示:与对照组相比,TNF-α+si-nc组和TNF-α+si-TAK1组H9C2心肌细胞活性显著降低(P>0.05);而与TNF-α+si-nc组相比,TNF-α+si-TAK1组H9C2心肌细胞活性显著升高(P>0.05)。运用流式细胞仪检测心肌细胞的细胞凋亡率,结果显示:与对照组相比,TNF-α+si-nc组和TNF-α+si-TAK1组H9C2心肌细胞活性显著升高(P>0.05);而与TNF-α+si-nc组相比,TNF-α+si-TAK1组H9C2心肌细胞活性显著降低(P>0.05),表5。此外,与TNF-α+si-nc组比较,TNF-α+si-TAK1组中caspase 3、8和9蛋白在H9C2心肌细胞表达显著降低(P<0.05),表6。

表5 TAK1敲低抑制TNF-α诱导的心肌细胞凋亡

表6 TAK1敲低对TNF-α诱导的心肌细胞凋亡相关蛋白表达的影响
2.5 TAK1敲低对TNF-α诱导的心肌细胞NF-κB信号通路的影响运用免疫印迹法检测NF-κB蛋白及其磷酸化情况,结果显示:三组间H9C2心肌细胞NF-κB蛋白表达无显著变化(图4);与TNF-α+si-nc组相比,TNF-α+si-TAK1组中p-NF-κB蛋白表达显著降低(P>0.05),表7。

表7 TAK1敲低抑制TNF-α诱导心肌细胞NF-κB信号通路的激活
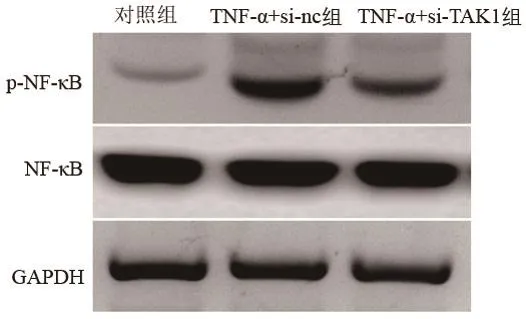
图4 TAK1敲低抑制TNF-α诱导的心肌细胞NF-κB信号通路的激活
2.6 TAK1过表达逆转miR-103a过表达对TNF-α诱导的心肌细胞凋亡的抑制作用运用CCK-8试剂盒检测H9C2心肌细胞的细胞活性,流式细胞仪检测细胞凋亡,结果显示:与TNF-α+miR-103a组相比,TNF-α+miR-103a+AAV-TAK1组H9C2心肌细胞细胞活性显著降低(P>0.05),而细胞凋亡率显著升高(P>0.05),表8。

表8 过表达TAK1逆转miR-103a过表达对TNF-α诱导的心肌细胞凋亡的抑制作用
2.7 TAK1过表达逆转miR-103a过表达对TNF-α诱导心肌细胞中NF-κB通路的抑制作用运用免疫印迹检测NF-κB蛋白及其磷酸化情况,结果显示:四组NF-κB蛋白表达无显著差异。而与TNF-α+miR-103a组相比,TNF-α+miR-103a+AAV-TAK1组H9C2心肌细胞p-NF-κB蛋白表达显著升高(P>0.05),图5及表9。

表9 TAK1过表达逆转miR-103a过表达对TNF-α诱导心肌细胞中NF-κB通路的抑制作用

图5 TAK1过表达逆转miR-103a过表达对TNF-α诱导心肌细胞中NF-κB通路的抑制作用
3 讨论
心血管疾病是严重威胁人类生命健康的一系列疾病的统称,心肌梗死、缺血心肌病、心力衰竭、心肌炎以及心律失常等都是常见的心血管疾病,这类疾病发病机制复杂,目前还未被完全揭示,但可明确的是炎症引起的心肌细胞损伤是心血管疾病主要的发病机制之一[1,2]。本研究使用TNF-α处理H9C2心肌细胞建立炎症诱导的心肌细胞损伤体外模型。TNF-α是一种由单核巨噬细胞、淋巴细胞以及粒细胞等分泌的细胞因子,不仅参与机体免疫调节,而且在炎症反应调控过程中发挥重要的抑炎作用[11,12]。研究表明[11,12],TNF-α是炎症诱导心肌损伤中重要的诱导因子,其不仅可以诱导心肌细胞凋亡,且会降低心肌细胞的增殖活性。本文研究发现,TNF-α处理的H9C2心肌细胞miR-103a表达降低,而TAK1蛋白表达升高,这与Chen[10]和Zhang等[13]的研究结果一致。Chen等[10]研究发现,缺氧诱导H9C2心肌细胞中miR-103a-3p表达降低;Zhang等[13]研究发现,TNF-α诱导滑膜成纤维样细胞miR-103a表达降低,而促进TAK1蛋白表达。进一步分析可知:转化生长因子β活化激酶 1(TAK1)被鉴定出是miR-103a的靶基因,而TAK1作为丝裂原活化蛋白激酶(MAP3K)家族的成员,是一种重要的NF-κB介导的炎症反应中的分子,可被多种炎症介质如TNF-α和 IL-1β通过MyD88/TRAFs 激活[14,15]。
miRNA是一种非编码小RNA,被发现在各种组织器官中广泛表达,并且由于其结构十分稳定、不易被降解而被认为是疾病诊断的优质生物标志物[3]。大量研究表明,miRNAs参与调控体内外心肌细胞的生存,比如H-F等[16]发现MiRNA-488-3p通过靶向ZNF791抑制急性心肌梗死诱导的心肌细胞凋亡;Li等[17]发现microRNA 340-5p通过调节Act1/NF来抑制缺氧/复氧诱导的心肌细胞凋亡和氧化应激。本研究发现,过表达miR-103a不仅可以抑制TNF-α诱导的H9C2心肌细胞凋亡,而且抑制TAK1蛋白表达,这与Zhang等的研究结果一致。Zhang等[13]在人滑膜成纤维样细胞中研究发现,miR-103a靶向抑制TAK1蛋白表达,并由此抑制炎症诱导的细胞损伤。
为进一步研究miR-103a是否通过靶向调控TAK1表达而诱导炎症环境下心肌细胞的凋亡,我们通过比对miR-103a和TAK1基因序列以查询其结合的部位,并通过双荧光素酶基因报告系统证实:miR-103在H9C2心肌细胞中靶向抑制TAK1表达。之前的研究表明,TAK1作为MAP3K家族的成员,是一种重要的炎症诱导因子,当其被TNF-α和IL-1β等炎症介质激活后,可通过磷酸化IKKs降解IKBα以促进NF-kB二聚体进入细胞核,最终促进炎症[14,15]。而本研究发现,敲低TAK1不仅抑制TNF-α诱导的H9C2心肌细胞凋亡,而且抑制NF-κB蛋白的磷酸化。NF-κB信号通路是一个不仅与细胞生长、增殖、凋亡以及分化等均密切相关,而且是与炎症调控密切相关的信号通路,其在多种心血管系统疾病中被过度激活[18,19]。研究指出,NF-κB蛋白被乙酰化或磷酸化修饰后进入细胞核是NF-κB信号通路被激活的重要标志,结合本文研究表明敲低TAK1可以通过抑制NF-κB信号通路的激活而减轻炎症诱导的心肌细胞凋亡。
此外,本文研究还发现,TAK1过表达不仅逆转miR-103a过表达对TNF-α诱导心肌细胞中NF-κB通路的抑制作用,而且逆转miR-103a过表达对TNF-α诱导心肌细胞中NF-κB通路的抑制作用。综上可知,TNF-α处理H9C2心肌细胞下调miR-103a和上调TAK1蛋白表达,miR-103a可通过靶向抑制TAK1而抑制NF-κB信号通路的激活,最终抑制TNF-α诱导的H9C2心肌细胞凋亡。
