Establishment of Molecular Biological Method for Identification of Bacteria by 16S rDNA and gyrB Gene
2023-05-16XiaxiaHOUYunxiaWANGCuizhiLIZhiyongLU
Xiaxia HOU, Yunxia WANG, Cuizhi LI, Zhiyong LU
Inner Mongolia Yili Industrial Group Co., Ltd., Hohhot 010110, China
Abstract [Objectives] The paper was to establish a molecular biological method for identification of bacterial strains. [Methods] The thalli of standard bacterial strains existing in the laboratory were collected and genomic DNA was extracted for amplification of 16S rDNA and gyrB gene. The 16S rDNA and gyrB gene sequences were obtained after sequencing. Sequences were aligned and analyzed via EzBioCloud and NCBI database, and phylogenetic trees were constructed to determine the species relationship of strains. Meantime, they were compared with known strains. [Results] This method could identify 5 standard strains accurately to the species level. The 16S rDNA and gyrB gene sequences were aligned and analyzed in EzBioCloud database and NCBI database. The strain with the max score was consistent with the known strain. And the query cover and ident were both above 99%. [Conclusions] The established molecular biological method for identification of bacterial strains by 16S rDNA and gyrB gene has good accuracy, which effectively solves the problem that the laboratory identification of bacteria relies on traditional methods and the accuracy can not be guaranteed, and further improves the identification ability of laboratory bacterial strains.
Key words 16S rDNA, gyrB, Bacterial identification, Molecular biological method
1 Introduction
Bacterial identification is mainly composed of phenotype and genotype identification[1-2]. Phenotype identification is the identification of bacteria through morphological observation, physiology and biochemistry using traditional culture methods. There are shortcomings such as long culture time, not obvious characteristic reaction, and easy to be affected by bacterial age, culture conditions and other external factors, so it is often difficult to achieve ideal identification effect for some difficult and rare bacteria[3-5]. Genotype identification is the application of molecular biological means in bacterial classification and identification, and strains tested are classified and identified at the molecular level through analysis of bacterial DNA. Common identification methods include gene sequencing[6-7], fingerprint technology[8-9], gene probe technology[10-11], polymerase chain reaction (PCR)[12-13],etc.Genotype identification is featured by high accuracy and good repeatability compared with traditional methods. At present, it is impossible to identify all bacterial strains with 100% accuracy through a single method, so it is necessary to combine morphological observation, biochemical identification, molecular biological identification and other means to accurately determine bacteria. Molecular biological identification is the "gold standard" for determination of bacteria[14].
Ribosomal DNA is the DNA sequence that encodes rRNA on biological chromosomes and exists in all biological chromosome genes. Ribosomal DNA-16S rDNA in prokaryotes is widely used in the identification of bacteria due to the existence of highly conserved regions and variable regions[15-16]. A highly conserved region refers to a specific region sequence that is the same in all bacteria, and a variable region refers to a specific region sequence that varies among different bacteria. The highly conserved region in 16S rDNA is used to design a universal primer for bacteria, further amplifies the variable region fragment in 16S rDNA of purified bacteria, and finally determines the species relationship of bacterial strains according to the sequencing and analysis of the variable region. In strain identification, the results of 16S sequence alignment are convincing at the genus level, so there are multiple results of the same genus but different species, which indicates that the experimental strain belongs to this genus. As for the species, it needs to be further identified through functional genes[17-18]. The functional genegyrB[19-20], with a single copy and a sequence length of 1.2-1.4 kb, is a common gene encoding gyrase B subunit in bacteria. This gene evolves quickly, with an average base replacement rate of 0.7%-0.8% per million years, which is faster than the 1% change in 16S rDNA per 50 million years, and can make up for the difficulty of accurately distinguishing bacteria sibling species by highly conserved 16S rDNA[21]. By establishing a molecular biological method for identification of bacteria by 16S rDNA andgyrB, this paper aimed to solve the problem that the laboratory bacteria identification relies on traditional methods and the accuracy can not be guaranteed, so as to further improve the identification ability of laboratory bacterial strains.
2 Materials and methods
2.1 Materials
2.1.1Standard strains and reagents. The standard strains used in the test includedPseudomonasaeruginosaATCC 27853,SalmonellaentericaCICC 22956,CronobactersakazakiiCICC 21560,EscherichiacoliATCC 25922, andStaphylococcusaureusATCC 25923.
The reagents used were universal primers for bacterial 16S rDNA, universal primers for bacterial functional genegyrB, small extraction kit for bacterial genomic DNA, PCR Mix premix with sample loading buffer (2×), agarose, DNA Marker (DL2000, 50×TAE buffer, 1×TAE buffer), GelRed dye (10 000×).
2.1.2Instrument and equipment. Biosafety cabinet, biochemical incubator, electric-heated thermostatic water bath, PCR meter, miniature centrifuge, electrophoresis apparatus, gel imager.
2.2 Methods
2.2.1Extraction of bacterial genome. The standard strains stored in the laboratory were selected and the thalli to be tested were collected. The genomic DNA was extracted according to the instructions of small extraction kit for bacterial genomic DNA.
2.2.2PCR amplification. Bacterial 16S rDNA sequence amplification was performed in a 50 μL reaction system containing 25 μL of PCR Mix premix, 1 μL of each 10 μmol/L primer 27 F and 1492 R, 22 μL of sterilized deionized water, and 1 μL of template. The sample template was the DNA of the strains extracted, the positive control template was the verified bacterial genome DNA, and the negative control template was sterilized deionized water. PCR amplification was conducted in the following procedures: pre-denaturating at 94 ℃ for 2 min; denaturating at 94 ℃ for 30 s, annealing at 55 ℃ for 90 s, extension at 72 ℃ for 1.5 min, 30 cycles; final extension at 72 ℃ for 10 min, preservation at 4 ℃. The amplification system of bacterial functional genegyrBsequence was the same as that of bacterial 16S rDNA sequence except for primersgyrBb-F andgyrBb-R. The reaction procedure was as follows: pre-denaturating at 94 ℃ for 2 min; denaturating at 94 ℃ for 30 s, annealing at 55 ℃ for 90 s, extension at 72 ℃ for 1 min, 30 cycles; final extension at 72 ℃ for 10 min, preservation at 4 ℃.
2.2.3Detection of PCR amplification products by electrophoresis. The amplified products were detected by electrophoresis with 1.5% agarose, with DL2000 DNA as the marker, and were imaged, observed and recorded by gel imager after electrophoresis.
2.2.4Sequencing of PCR amplification products of samples to be identified. If the bacterial genome extraction and PCR reaction were normal, the PCR amplification products of the samples to be identified would be sent to Sangon Biotech (Shanghai) Co., Ltd. for sequencing.
2.2.5Sequencing splicing and analysis of PCR amplification products. Observation of sequencing peak map: Professional software BioEdit was used to observe the sequencing peak chart to determine whether the sequencing results were normal. If the entire sequence peak chart had overlapping peaks, it indicated that the bacteria to be identified had not been completely isolated and purified, and there was interference from infectious bacteria, or pollution brought into the PCR process affected the sequencing result. At this time, the sequencing result was judged to be abnormal and discarded. Splicing and alignment analysis of sequencing results: Professional software DNAMAN was used to splice the two sequences, and the spliced sequences were analyzed via online analysis websites NCBI and EzBioCloud.
2.2.6Construction of phylogenetic tree. Collection of strain 16S rDNA sequence: According to the analysis results in Section2.2.5, the 16S rDNA sequences of related species were downloaded from EzBioCloud website and sorted into a file, which was then saved in FASTA format. Construction of phylogenetic tree: Professional software MEGA was used to align and analyze the collated sequences, and Neighbor-Joining phylogenetic trees were constructed after removing the redundant sequences at both ends.
2.2.7Result interpretation. The detection results of 16S rDNA sequence should be interpreted first. When the detection results of 16S rDNA sequence could not accurately identify the strain to the species level, the detection results ofgyrBsequence should be interpreted. In the interpretation of 16S rDNA sequence results, EzBioCloud and NCBI alignment results and 16S rDNA phylogenetic tree results should be integrated.
3 Results and analysis
After DNA extraction, 16S rDNA sequence andgyrBsequence of strains tested were amplified. After detected by electrophoresis, obvious and bright amplification bands appeared at 1 500 bp and 1 200 bp, which could be judged that the genome extraction, 16S rDNA PCR assay andgyrBPCR assay of the bacteria to be identified were normal. Subsequently, the sequencing results of the strains tested were analyzed. MEGA7 software was used to build phylogenetic trees, and the evolutionary distance was calculated using Kimura 2-parameter method. The 16S rDNA sequence phylogenetic trees of various strains are shown in Fig.1-5, and only values with probability greater than 70% are shown in the figures.
3.1P.aeruginosaThe 16S rDNA sequence of this strain was aligned and analyzed in EzBioCloud database, and the alignment result showed that onlyP.aeruginosahad the similarity greater than 98.7%, so the strain was identified to beP.aeruginosa. The 16S rDNA sequences were aligned and analyzed in NCBI database. The top 5 strains with the max scores wereP.aeruginosa, and the query cover and ident were 100%. As can be seen from the phylogenetic tree in Fig.1, the evolutionary distance betweenP.aeruginosaand the strain was closest. In summary, the strain was identified to beP.aeruginosa. The known standard strain wasP.aeruginosa, indicating that the method was accurate and could identifyP.aeruginosato the species level.

Fig.1 16S rDNA phylogenetic tree of Pseudomonas aeruginosa
3.2S.entericaThe 16S rDNA sequence of this strain was aligned and analyzed in EzBioCloud database, and the alignment result showed that onlyS.entericahad the similarity greater than 98.7%, so the strain was identified to beS.enterica. The 16S rDNA sequences were aligned and analyzed in NCBI database. The top 5 strains with the max scores were allS.enterica, and the query cover and ident were 100%. As can be seen from the phylogenetic tree in Fig.2, the strain had the closest evolutionary distance withS.enterica. In summary, the strain was identified to beS.enterica. The known standard strain wasS.enterica, indicating that the method was accurate and could identifyS.entericato the species level.
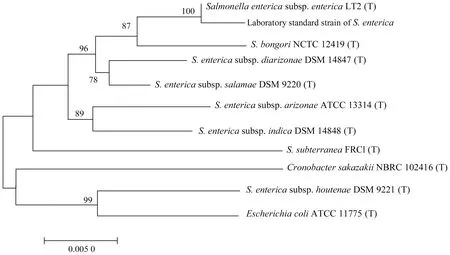
Fig.2 16S rDNA phylogenetic tree of Salmonella enterica
3.3C.sakazakiiThe 16S rDNA sequences were aligned and analyzed in EzBioCloud database, and there were three species of the genusCronobacterwith the similarity greater than 98.7%. Therefore, the strain was identified to beCronobacter. The 16S rDNA sequences were aligned and analyzed in NCBI database. The top 5 strains with the max scores wereC.sakazakii, and the query cover and ident were above 99%. According to the phylogenetic tree in Fig.3, it can be seen that the strain had the closest evolutionary distance withC.sakazakiiandC.malonaticus. Partial sequences ofgyrBgene of the strain were aligned withgyrBgene ofCronobacterstrains in NCBI database. Compared withC.sakazakii, the query cover was 100% and the ident was more than 99%, and the alignment score was 2 126. Compared with otherCronobacterspecies, the ident was as high as 96%, so the identification result ofgyrBgene confirmed that the strain wasC.sakazakii. In summary, the strain was identified to beC.sakazakii. The known standard strain wasC.sakazakii, indicating that the method was accurate and could accurately identifyC.sakazakiito the species level.
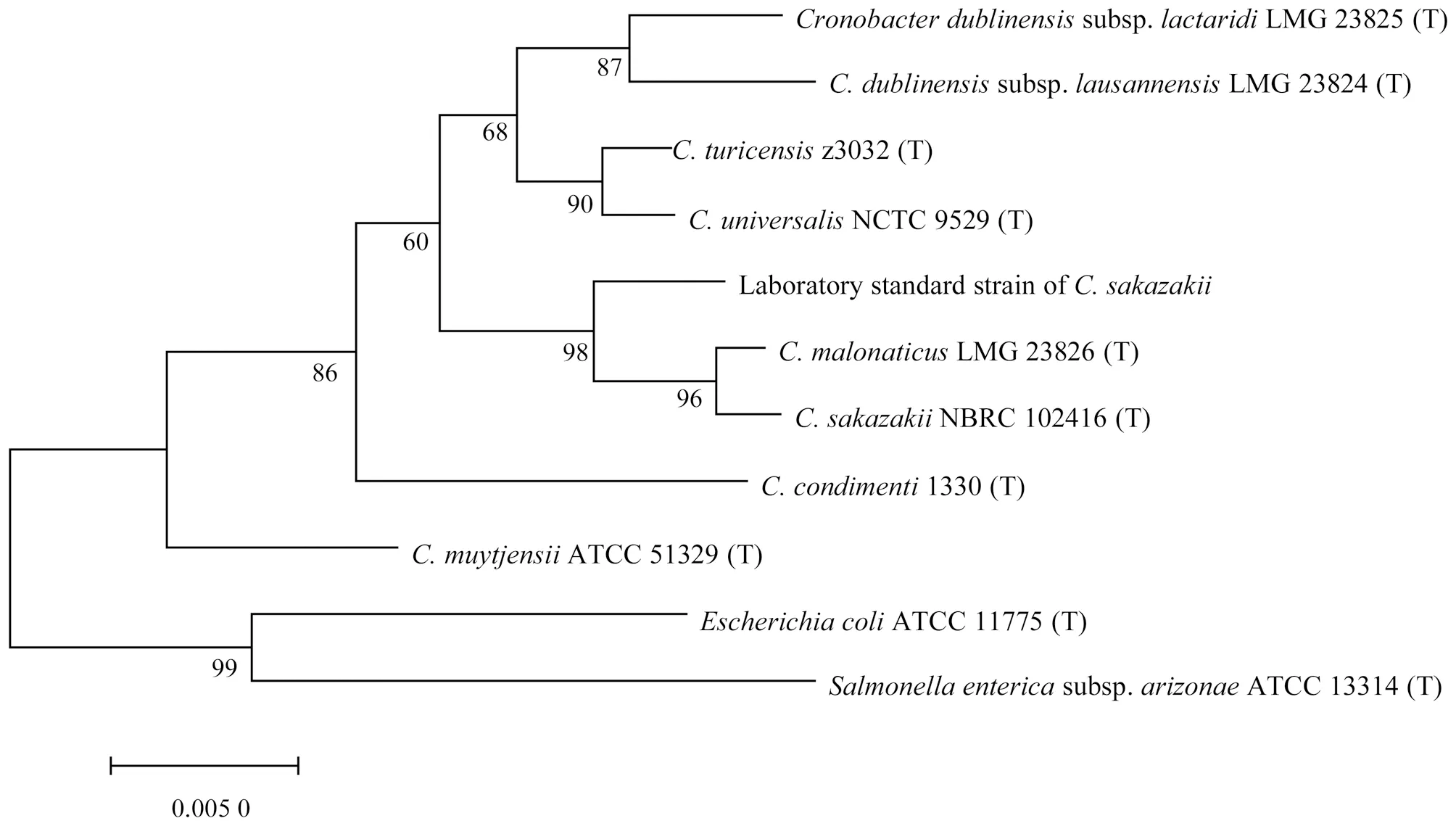
Fig.3 16S rDNA phylogenetic tree of Cronobacter sakazakii
3.4E.coliThe 16S rDNA sequences were aligned and analyzed in EzBioCloud database. The alignment results showed that 4 strains ofShigellaand 4 strains ofEscherichiahad the similarity greater than 98.7%, which were identified to be Enterobacteriaceae. The 16S rDNA sequences were aligned and analyzed in NCBI database. The top 5 strains with the max scores wereE.coli, and the query cover and ident were more than 99%. As can be seen from the phylogenetic tree in Fig.4, the strain had the closest evolutionary distance withE.coli. Partial sequences ofgyrBgene of this strain were aligned with those ofShigellaandEscherichiastrains in NCBI database. The highest query cover and ident were more than 99%, but the score ofE.coliwas higher than that ofShigella. In summary, the strain was identified to beE.coli. The known standard strain belonged toE.coli, indicating that the method was accurate and could accurately identifyE.colito the species level.
3.5S.aureusThe 16S rDNA sequences were aligned and analyzed in EzBioCloud database, and there were 4 strains ofStaphylococcuswith the similarity greater than 98.7%, so the strain was identified to beStaphylococcus. The 16S rDNA sequences were aligned and analyzed in NCBI database. The top 5 strains with the max scores were allS.aureus, and the query cover and ident were 100%. As can be seen from the phylogenetic tree in Fig.5, the strain had the closest evolutionary distance withS.aureus. Partial sequences ofgyrBgene of this strain were aligned with those ofStaphylococcusstrains in NCBI database. Compared withS.aureus, the query cover was 100%, the ident was more than 99%, and the alignment score was 2 162. Compared with otherStaphylococcusstrains, the highest ident was 95%, so the identification result ofgyrBgene confirmed that the strain wasS.aureus. In conclusion, the strain was identified to beS.aureus. The known standard strain wasS.aureus, indicating that the method was accurate and could accurately identifyS.aureusto the species level.

Fig.5 16S rDNA phylogenetic tree of Staphylococcus aureus
4 Discussion
Molecular biological methods to some extent make up for the shortcomings of traditional methods such as long identification cycle, easy to be affected by culture conditions, poor repeatability and low accuracy, and have fast identification and high accuracy when exploring the taxonomic status and species relationship of bacteria from the gene level. In the classification of species, if the homology of 16S rDNA sequence between two taxonomic units is higher than 97.5%, the two taxonomic units can be classified as the same species[22]. However, for some closely related strains, there may be more than one strain with high homology after alignment of 16S rDNA gene sequence in database, so it can not be differentiated at the species level, and other methods should be combined to supplement identification.
Recent studies have found that thatgyrAandgyrBgenes are widely used because they can make up for the shortcomings of 16S rDNA gene method. The combined identification method of 16S rDNA andgyrBgene sequence is a scientific and internationally recognized method for identification of newly isolated species[23]. At present, most studies have applied the combination method of 16S rDNA andgyrBgene in the identification of closely related bacterial populations[24-26], and received good results. Compared with traditional methods and single use of 16S rDNA sequence for identification, the method can identify strains quickly and accurately to the species level. Fu Qietal.[27-28]used a phylogenetic tree based on 16S andgyrBgene sequences to accurately and rapidly identify a strain ofBacilluslicheniformisisolated from seawater and a strain ofB.amyloliquefaciensisolated from soil. Lu Jiaqietal.[29]used combined 16S rDNA andgyrBgene sequence to identify a strain ofB.cereus.
5 Conclusions
In this study, a molecular biological method of combined 16S rDNA andgyrBidentification of bacterial strains was established. Five standard strains, which were the most representative in the laboratory, were selected for experiments. Proper amount of thalli were collected to extract genomic DNA, and 16S rDNA andgyrBgene sequences were obtained through 16S rDNA amplification andgyrBgene sequencing. The 16S rDNA sequences were aligned and analyzed in EzBioCloud database and NCBI database, and the phylogenetic trees were constructed.P.aeruginosaandS.typhimuriumcould be accurately identified to the species level by 16S rDNA sequence analysis. ButC.sakazakii,E.coliandS.aureuscould only be identified to the genus level through 16S rDNA sequence analysis. Therefore, continued interpretation ofgyrBsequence detection results finally accurately identified the strain to the species level. The results were consistent with known standard strains, indicating that this method had good accuracy, andgyrBgene could make up for the deficiency of 16S rDNA molecular identification method.
杂志排行

Asian Agricultural Research的其它文章
- Analysis of Visualization Mapping in the Field of Brand Community Based on CiteSpace III
- Amplification and Bioinformatics Analysis of h-ns Gene of Vibrio alginolyticus
- Cloning and Bioinformatics Analysis of vscB Gene of T3SS Chaperone of Vibrio alginolyticus
- Effects of Continuous Cropping and Crop Rotation on Fungal Diversity in Greenhouse Soil
- Effects of Addition of Amino Acids or Soybean Phospholipid in Diet on Slaughter Performance and Meat Quality of Pigs
- Changes in Soil Organic Matter, Nitrogen and Phosphorus Contents during Decomposition of Pear Branches