杨梅根药材质量标准研究*
2023-05-11耿冶飞陈秀杰郑风敏王永杰陶晓赛
耿冶飞,陈秀杰,郑风敏,王永杰,陶晓赛
(河南省鹤壁市食品药品检验检测中心,河南 鹤壁 458030)
杨梅根为杨梅科植物矮杨梅Myrica nanaCheval.的干燥根,产于云南中部,向东达贵州西部,主要生长于海拔1 500~3 500 m 的山坡、林缘及灌木丛中[1]。矮杨梅为云南地方习用药材,以根皮、茎皮、果实入药,《云南中草药》、《中药大辞典(下册)》[2]、《中华本草(第二册)》[3]、《云南省中药材标准(第一册)》[4]、《卫生部药品标准 中药成方制剂》[5-7]中均有收载。其根皮味涩、性凉,果实味酸、性凉,具有收敛、止血、消炎功效,主治痢疾、泄泻、脱肛、崩漏、消化道出血、风湿疼痛、外伤出血等症。杨梅根主要含有黄酮及其苷类、二芳基庚烷类、三萜及其苷类、没食子酸、β-谷甾醇、鞣质等成分,其中分离最多的黄酮类化合物为杨梅苷。药理学研究表明,黄酮类化合物具有祛痰、止咳、对抗自由基、抗癌、防癌、抗菌、消炎、防治心脑血管疾病等作用[8-13]。为控制杨梅根药材的质量,本研究中通过药材性状、粉末显微特征和薄层色谱对其进行定性鉴别;通过测定其水分、总灰分、酸不溶性灰分、醇溶性浸出物,初步拟订检查项和浸出物限量;并选择化学结构明确、含量较高的杨梅苷作为指标性成分,建立了测定其含量的高效液相色谱法。现报道如下。
1 仪器与试药
1.1 仪器
N - 300M 型生物显微镜(宁波永新光学股份有限公司),配有显微成像系统;Nanomat 4 型薄层色谱手动点样仪(瑞士卡玛公司);AE240 型电子分析天平(梅特勒-托利多仪器<上海>有限公司,精密度为0.1 mg);GoodLook - 1000 型薄层色谱成像系统(上海科哲生化科技有限公司);DHG-9145A型电热鼓风干燥箱,SX2-4-10型箱式电阻炉,均购自上海一恒科学仪器有限公司;SK2510HP 型超声波清洗机(上海科导超声仪器有限公司,功率为250 W,频率为53 kHz);e2695型高效液相色谱仪(美国Waters公司)。
1.2 试药
杨梅苷对照品(中国食品药品检定研究院,批号为111860-201703,含量以95.2%计);聚酰胺薄膜(浙江省台州市路桥四甲生化塑料厂);甲醇、乙腈(色谱纯,霍尼韦尔贸易<上海>有限公司);水为娃哈哈纯净水,其余试剂均为分析纯;16 批杨梅根(编号为S1 -S16)采自云南省不同地区,经河南省鹤壁市食品药品检验检测中心陈秀杰副主任药师鉴定为正品,采集后均按试验要求洗净、晾干,备用。样品信息见表1。
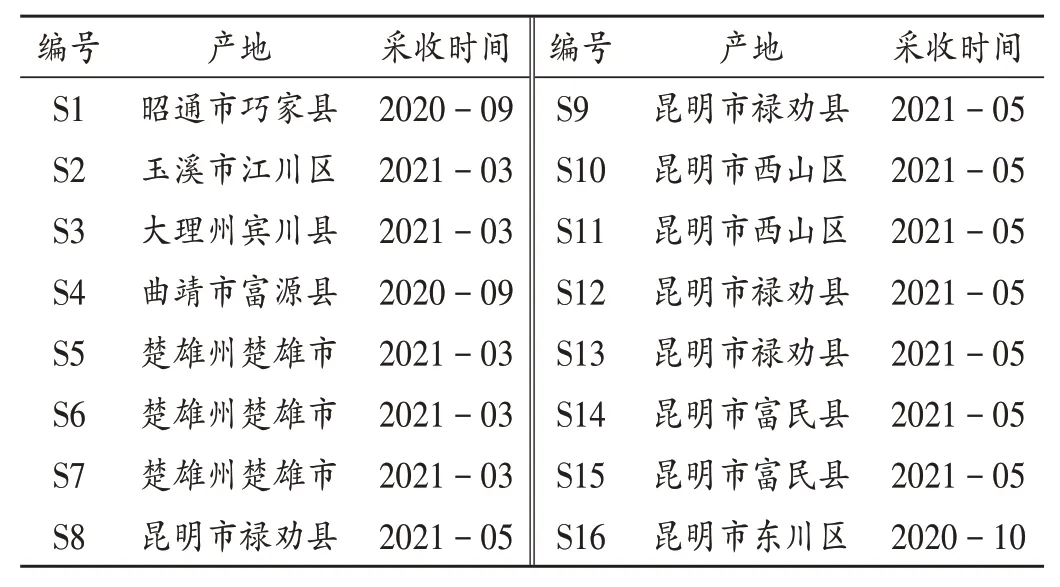
表1 杨梅根样品信息Tab.1 Information of Myricae Radix samples
2 方法与结果
2.1 性状
杨梅根药材呈圆柱形,粗壮扭曲,多有分枝,直径0.5~8.0 cm。表面褐色,略粗糙,有纵皱纹及横形皮孔样痕迹。质硬而韧,难折断,断面不平坦,皮部狭窄,深棕色,木部宽广,浅棕色至棕色,有细密的放射状纹理。气微,味微涩。
2.2 鉴别
显微鉴别:取药材粉末(过4 号筛)少许,置载玻片上,制片,显微镜下观察。粉末呈灰棕色。木栓细胞呈淡黄色,呈类长方形或多角形,壁厚。韧皮纤维末端较钝圆,直径15~23 μm,壁厚,胞腔极窄,孔沟较密,有的深达纤维壁,胞腔内可见点状纹孔。木纤维散在或成束,纤维末端较尖,直径11~22 μm,壁厚,胞腔具稀疏斜纹孔。石细胞呈类椭圆形或类长方形,单个散在或多个集聚,直径26~89 μm,壁厚,孔沟明显,胞腔内有黄棕色物或具点状纹孔。草酸钙方晶较多,散在,呈方形或不规则多面体,直径13~22 μm。导管多为网纹导管,网眼排列较紧密,少数为具缘纹孔导管,直径11~37 μm。详见图1。

图1 杨梅根粉末显微特征图1.Cork cells 2.Phloem fiber 3.Wood fiber 4.Stone cell 5.Calcium oxalate square crystal 6.Reticulated vessel 6′.Bordered pit vesselFig.1 Microscopic characteristics of Myricae Radix powder
薄层色谱鉴别:取药材粉末0.5 g,置具塞锥形瓶中,加乙酸乙酯10 mL,超声处理10 min,滤过,滤液作为供试品溶液。另取杨梅苷对照品,加乙酸乙酯制成每1 mL 含0.5 mg 的溶液,作为对照品溶液。按2020年版《中国药典(四部)》通则0502 薄层色谱法试验,吸取上述2 种溶液各1 μL,分别点于同一聚酰胺薄膜上,以乙醇-水-冰乙酸(6∶4∶1,V/V/V)为展开剂,展开,取岀,晾干,喷以三氯化铝试液,置紫外光灯(365 nm)下检视。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的荧光斑点。色谱图见图2。

图2 杨梅根薄层色谱图(365 nm)S.Reference solution 1-16.Test solution(No.S1-S16)Fig.2 TLC chromatograms of Myricae Radix(365 nm)
2.3 检查
水分:取16批药材样品粉末,按2020年版《中国药典(四部)》通则0832 水分测定法第二法(烘干法)测定,结果见表2。根据测定结果,水分拟订为不得过10.0%。

表2 16批杨梅根药材检查项和其中杨梅苷含量测定结果(%)Tab.2 Inspection items of 16 batches of Myricae Radix and results of the content determination of myricetin in Myricae Radix(%)
总灰分:取16 批药材样品粉末,按2020年版《中国药典(四部)》通则2302 灰分测定法测定,结果见表2。根据测定结果,总灰分拟订为不得过5.0%。
酸不溶性灰分:取16批药材样品粉末,按2020年版《中国药典(四部)》通则2302 灰分测定法测定,结果见表2。根据测定结果,酸不溶性灰分拟订为不得过2.0%。
醇溶性浸出物:取16 批药材样品粉末,按2020年版《中国药典(四部)》通则2201 浸出物测定法项下的冷浸法,以60%乙醇为溶剂测定,结果见表2。根据测定结果,醇溶性浸出物拟订为不得少于15.0%。
2.4 含量测定
2.4.1 色谱条件与系统适用性试验
色谱柱:Symmetry®C8柱(250 mm×4.6 mm,5 μm);流动相:乙腈-0.1%磷酸水溶液(13∶87,V/V);流速:1.0 mL/ min;检测波长:260 nm;柱温:40 ℃;进样量:5 μL。对照品溶液、供试品溶液、空白溶液在此色谱条件下的色谱图见图3。可见,理论板数按杨梅苷峰计不低于3 000,杨梅苷色谱峰均达到基线分离,分离度均大于1.5,且空白溶液对测定无干扰。

图3 高效液相色谱图1.MyricetinA.Reference solution B.Test solution C.Blank solutionFig.3 HPLC chromatograms
2.4.2 溶液制备
取杨梅苷对照品适量,精密称定,加甲醇制成每1 mL含0.1 mg 的对照品溶液。取药材粉末(过3 号筛)0.3 g,精密称定,置具塞锥形瓶中,精密加入甲醇50 mL,密塞,称定质量,放置30 min,超声处理(功率为250 W,频率为53 kHz)45 min,放冷,再称定质量,用甲醇补足减失的质量,摇匀,滤过,取续滤液,即得供试品溶液。以甲醇为空白溶液。
2.4.3 方法学考察
线性关系考察:取杨梅苷对照品适量,精密称定,加甲醇分别制成每1 mL 含杨梅苷0.01,0.025,0.05,0.1,0.15,0.2,0.25 mg的系列标准溶液,按2.4.1项下色谱条件进样测定,以杨梅苷质量浓度(X,mg/mL)为横坐标、峰面积为纵坐标(Y)进行线性回归,得回归方程Y=107X-6 086.2(r=1.000 0,n=7)。结果表明,杨梅苷质量浓度在0.01~0.25 mg/ mL 范围内与峰面积线性关系良好。
检测限与定量限确定:取2.4.2 项下对照品溶液适量,加甲醇稀释成系列质量浓度,按2.4.1 项下色谱条件进样测定,记录色谱图,以信噪比(S/N)为3∶1 时的质量浓度为检测限,以S/N为10∶1 时的质量浓度为定量限。结果检测限为0.002 1 mg/ mL,定量限为0.007 0 mg/mL。
精密度试验:取2.4.2 项下对照品溶液适量,按2.4.1 项下色谱条件连续进样测定6 次,记录色谱峰峰面积,结果杨梅苷峰面积的RSD为0.52%(n=6),表明仪器日内精密度良好。取上述对照品溶液每天进样2次,连续测定3 d,记录色谱峰峰面积,结果杨梅苷峰面积的RSD为1.39%(n=6),表明仪器日间精密度良好。
重复性试验:取药材样品(编号为S8)粉末6 份,每份0.3 g,精密称定,按2.4.2 项下方法制备供试品溶液,按2.4.1项下色谱条件进样测定,记录峰面积,并计算杨梅苷含量。结果杨梅苷含量的RSD为0.58%(n=6),表明方法重复性良好。
稳定性试验:取2.4.2 项下供试品溶液适量,分别于0,4,8,12,18,24,36,48 h 时按2.4.1 项下色谱条件进样测定,记录峰面积。结果杨梅苷峰面积的RSD为1.36%(n=8),表明供试品溶液在48 h内稳定性良好。
加样回收试验:取已知杨梅苷含量的药材样品(编号为S8)粉末6 份,每份0.15 g,精密称定,分别加入2.4.2 项下对照品溶液适量,依法制备供试品溶液,按2.4.1 项下色谱条件进样测定,记录峰面积,并计算加样回收率。结果见表3。
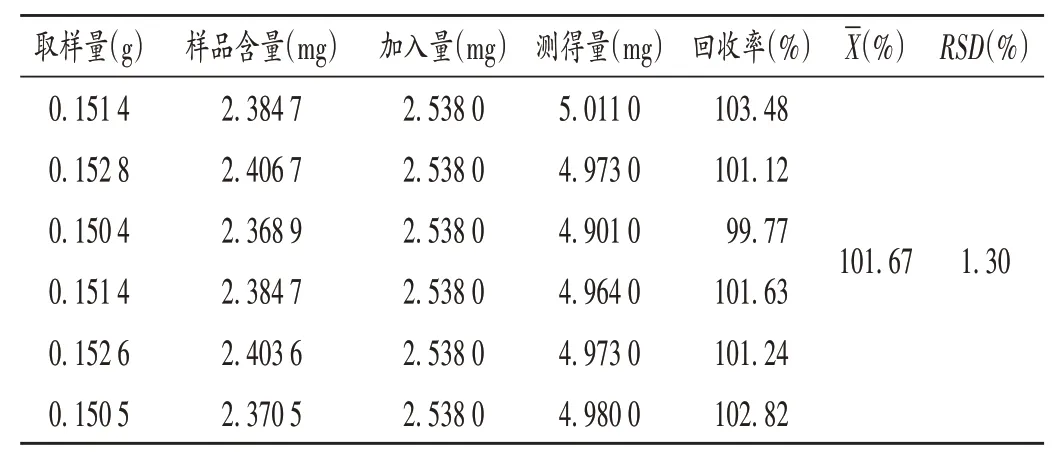
表3 加样回收试验结果(n=6)Tab.3 Results of the recovery test(n=6)
2.4.4 样品含量测定
分别取16批(编号为S1-S16)药材样品粉末0.3 g,精密称定,按2.4.2项下方法制备供试品溶液,按2.4.1项下色谱条件进样测定,记录峰面积,并计算杨梅苷含量。结果见表2。根据测定结果,拟订杨梅苷含量按干燥品计算不得少于1.5%。
3 讨论
3.1 薄层色谱条件选择
根据杨梅根药材中所含化学成分,选择其所含化学成分较高的黄酮类化合物杨梅苷作为指标性成分对其进行薄层色谱鉴别。杨梅苷为黄酮醇苷类,可用乙酸乙酯、丙酮、乙醇、甲醇等溶剂提取。本研究中参考文献[4,14],分别以乙酸乙酯、甲醇、60%乙醇为溶剂进行提取,结果以甲醇和60%乙醇为提取溶剂时,色谱图斑点较多,且有杂质斑点干扰;以乙酸乙酯为提取溶剂时主斑点清晰,且无杂质斑点干扰。故选择乙酸乙酯作为提取溶剂。
采用硅胶G 薄层板时,分别以三氯甲烷- 甲醇-甲酸(20∶5∶2,V/V/V)和乙酸乙酯-甲酸-水(8∶1∶1,V/V/V)为展开剂进行试验,结果薄层色谱的比移值偏低,且斑点分离度不理想;采用聚酰胺薄膜时,分别以异丙醇- 水(3∶2,V/V)和甲醇- 水- 冰乙酸(4∶5∶1,V/V/V)为展开剂进行试验,结果薄层色谱主斑点的比移值较合适,且斑点较清晰。故选择聚酰胺薄膜作为固定相。当采用聚酰胺薄膜时,用以上2 种展开剂的展开时间较长,且主斑点有拖尾现象,参考文献[15],分别采用正丁醇- 乙醇- 水(2∶4∶5,V/V/V),乙醇- 水-冰乙酸(7∶3∶1,V/V/V),乙醇- 水- 冰乙酸(6∶4∶1,V/V/V)为展开剂进行试验,结果以乙醇-水-冰乙酸(6∶4∶1,V/V/V)为展开剂时的展开效果最好。故选择此溶剂为展开剂。
关于薄层色谱点样量的确定,分别用对照品溶液和供试品溶液点样1,2,5,10 μL 于同一聚酰胺薄膜上进行比较,结果采用聚酰胺薄膜时,点样量不宜过大,当点样量超过2 μL 时主斑点有拖尾现象,且点样量越大,拖尾现象越明显。故选择点样量为1 μL。
3.2 浸出物测定条件选择
杨梅根所含成分为黄酮类化合物,故选择不同体积分数的乙醇提取较合适。本研究中按2020年版《中国药典(四部)》通则2201 醇溶性浸出物测定法项下的冷浸法和热浸法,分别用无水乙醇、75%乙醇、60%乙醇、50%乙醇作为溶剂,选定3 批样品进行测定,结果在相同体积分数乙醇条件下,热浸法浸出物含量高于冷浸法浸出物含量。在不同体积分数乙醇条件下,60%乙醇和50%乙醇浸出物含量高于无水乙醇和75%乙醇浸出物含量。比较冷浸法60%乙醇和50%乙醇浸出物含量,发现60%乙醇浸出物含量稍高于50%乙醇浸出物含量。经综合考虑,最终选择60%乙醇为溶剂,采用冷浸法测定药材中的浸出物。
3.3 含量测定条件选择
参考文献[4],本研究中考察了以甲醇作为溶剂,分别采用加热回流提取1 h 和2 h,索氏抽提1,2,3 h,超声提取30,45,60 min 等不同提取方法,发现以索氏抽提法和超声提取法的提取效率较高,考虑到超声提取法较简便,更易于操作,最终选择超声提取法进行提取。试验发现,超声提取45 min 和60 min 的提取效率相当,为节约时间,最终选择超声提取45 min 作为提取条件。
对杨梅苷对照品溶液进行全波长扫描(190~400 nm),结果显示,于260 nm波长处有较大吸收。还考察了甲醇-水溶液、乙腈-水溶液、甲醇-0.1%磷酸水溶液、乙腈-0.1%磷酸水溶液[4]、乙腈- 0.5%磷酸水溶液[14]等不同流动相系统,分别选择不同规格的C18柱、C8柱,设置不同色谱柱温度(25,30,35,40 ℃)对色谱条件进行考察。试验发现,当色谱柱选择C18柱时,有杂质峰影响杨梅苷色谱峰的分离,而C8柱则无杂质峰干扰,故选择色谱柱为C8柱;当流动相为乙腈-0.1%磷酸水溶液(13∶87,V/V),色谱柱温度为40 ℃时,色谱峰的对称性和分离度均较好,且出峰时间适宜,无杂质峰干扰,故确定2.4.1项下色谱条件。
3.4 限度指标拟订
根据试验结果,将水分的平均值上浮30%,拟订水分限度标准;将醇溶性浸出物的平均值下浮30%,拟订浸出物限度标准;将含量测定结果平均值下浮25%,拟订含量限度标准;考虑到实际采收、加工、生产等情况对总灰分和酸不溶性灰分造成的影响,参考一般根及根茎类药材总灰分和酸不溶性灰分的限度标准。初步拟订杨梅根药材质量标准为,水分不得过10.0%,总灰分不得过5.0%,酸不溶性灰分不得过2.0%,醇溶性浸出物含量不得少于15.0%,杨梅苷含量按干燥品计算不得少于1.5%。
