儿童Kearns-Sayre综合征心电图改变及心脏性猝死的预防
2023-05-08高春梅陈烨刘影沈洁黎旋王波张帆束文娟孙凌吕海涛
高春梅 陈烨 刘影 沈洁 黎旋 王波 张帆 束文娟 孙凌 吕海涛
Kearns-Sayre综合征是一种线粒体疾病,由于线粒体DNA(mitochondrial DNA,mtDNA)缺陷,导致三磷酸腺苷(ATP)合成障碍,造成能量来源不足的一种异质性疾病,可累及全身各个系统,尤以心脏传导系统的病变最为严重,导致完全性房室传导阻滞、室性心动过速(简称室速)、心室颤动(简称室颤)等致命性心律失常,发生心脏性猝死。由于该病较为罕见,临床对该病认识有限,极易漏诊、误诊。笔者回顾分析苏州大学附属儿童医院心内科诊治的1 例及文献检索的21例Kearns-Sayre综合征患儿,总结其临床、心电图动态变化情况、治疗及随访情况,为预警心脏性猝死,确定起搏器或埋藏式心脏转复除颤器(ICD)的植入时机提供参考。
1 资料与方法
1.1 临床资料患儿男性,12岁,因“运动后头晕、胸闷3年余,加重2月余”入住本院心内科。心电图示窦性心律、完全性右束支阻滞、间歇性左前分支阻滞(图1);运动后复查心电图示窦性心律、完全右束支阻滞并左前分支阻滞、一度房室传导阻滞(图2),且QRS波形态多变。追问病史,患儿2个月前外院查心电图提示完全性右束支阻滞(未见报告)。动态心电图示:窦性心律、窦性心律不齐、部分时段窦性心动过缓(平均心率75次/分,最小心率45次/分,最大心率117次/分,最长RR 间期1 388 ms)、完全性右束支阻滞、间歇性左前分支阻滞、间歇性一度房室传导阻滞、偶见室上性早搏。尿常规:蛋白:50 mg/dl(+);血常规、生化全套、血气加电解质、心肌酶谱、甲状腺功能全套、血沉、抗O、HLA-B27、心脏彩色超声心动图、胸部CT 均未见明显异常。患儿产前及出生史均正常,入院前3年曾诊断为矮小症,身材比例正常,生长激素治疗两年,第一年生长5 cm,第二年未再生长,故停药。母亲有“强直性脊柱炎”及“糖尿病”史,父亲体健,非近亲结婚。完善检查后患儿办理出院,定期门诊随访。
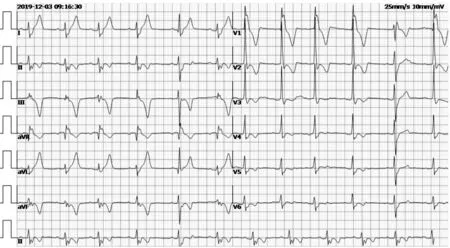
图1 患者入院时常规心电图
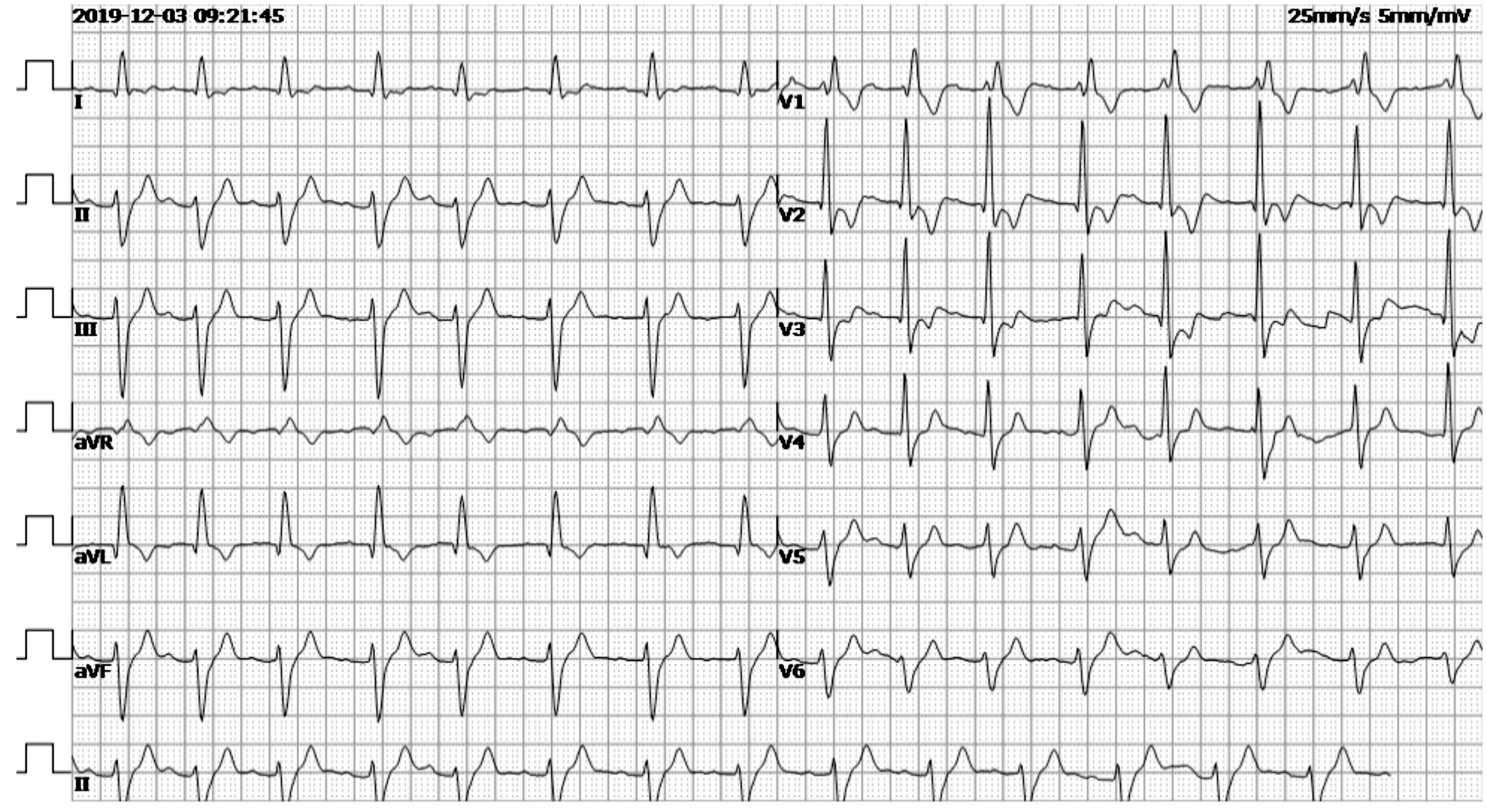
图2 患儿运动后记录的常规心电图
患儿出院后1周、3个月时复查常规心电图较前相仿。于出院后1年1个月复查动态心电图示窦性心律、窦性心律不齐、部分时段窦性心动过缓(平均心率46 次/分,最小心率34 次/分,最大心率63次/分,最长RR 间期1 920 ms)、完全性右束支阻滞、间歇性左前分支阻滞、部分时段可见二度2∶1房室传导阻滞。提示疾病较前进展。因患儿家长拒绝完善基因检查,故嘱其定期门诊随访。
患儿于出院后1年6个月时因“头晕伴视力下降”再次入院治疗,行头颅MRI示两侧大脑半球白质、内囊、大脑中脑脚、脑桥、延髓弥漫性脱髓鞘病变;双侧眼球MRI未见明显异常;两侧视神经远端异常信号;双侧上颌窦、筛窦及左侧蝶窦炎。眼底检查:双眼底视网膜可见骨细胞样色素沉着,考虑视网膜色素变性;视野检查:双眼颞侧视野缺损。脑电图及脑电地形图未见明显异常;泌乳素升高,为29.16 ng/ml;脑脊液检查示蛋白为150 mg/dl(>100 mg/dl)。对患儿行线粒体基因组测序发现了chr M:6341-13993的缺失。对该结果进行了长程PCR 验证,结果如图3所示。体格检查:眼睑下垂,眼球活动受限,不能内聚,不能向上、向下活动,指鼻试验、跟-膝-胫试验阳性。结合患儿存在心脏传导系统阻滞、矮小等病史,“Kearns-Sayre综合征”诊断明确。予鸡尾酒疗法(维生素A、维生素E、维生素B1、维生素B2)及艾地苯醌、中成药(易谷丹、护心丸)口服后予出院。

图3 血液样本的线粒体长程PCR 结果
出院后患儿仍有头晕、恶心及乏力,视力降低,爬楼梯费力,运动耐力下降。出院后第3个月共出现6次“晕厥”,又再次住院治疗查长程脑电图提示:正常范围脑电图;脑地形图可见前头部慢频带增高;肌电图未见明显异常;附见心律不齐:清醒期心率约40~60次/分,睡眠期心率约20~40次/分,睡眠期检见十余次停博,最长约10 s。常规心电图:窦性心律,三度房室传导阻滞(心房率88次/分、心室率36次/分),室性逸搏心律(图4)。动态心电图:窦性心动过缓(平均心率42次/分,最小心率23次/分,最大心率60次/分,有31次大于2.0 s的停搏,其中最长为7.12 s)、二度至三度房室传导阻滞、完全性右束支阻滞合并左前分支阻滞、心室停搏、室性逸搏心律。入院后第6天在全麻下行双腔永久起搏器置入术,选择美敦力植入式心脏起搏器(程控设置起搏器DDDR 模式,起搏频率60次/分,起搏电压3.5 V)后出院(图5),门诊定期随访。期间患儿QTc一直处于正常范围。
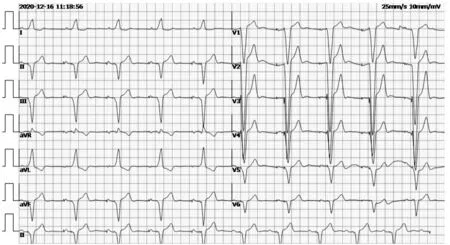
图5 患儿起搏器植入术后心电图
1.2 文献检索文献检索儿童Kearns-Sayre综合征21例[1-6]和本例患儿1例,共22例。对22例患儿进行临床资料分析。
2 结果
2.1 临床表现22例患儿中,男13例,女9例,发病年龄2.5~13岁,中位年龄8.5岁,其中起病表现以上睑下垂10例,眼肌麻痹5例,晕厥3例,视力下降1例,生长迟缓2例,四肢瘫痪1例。临床表现各不相同,主要表现为上睑下垂、眼肌麻痹、视网膜色素变性、共济失调、生长迟缓、肌无力、视力下降、晕厥等,也有少部分患儿出现糖尿病、耳聋、慢性肾脏疾病等症状。所有患儿均无家族相关遗传病史。
2.2 心电图表现及实验室检查所有患儿首次心电图检查均出现不同程度的传导阻滞:①11 例(50%)患儿入院时表现为单纯左前分支阻滞,有9例患儿进展为左前分支阻滞合并完全性右束支阻滞(潜伏期1~7年);②3例(13.7%)患儿入院时表现为单纯完全性右束支阻滞(其中1例合并电轴左偏及低电压),1例进展为左前分支阻滞合并完全性右束支阻滞;③6例(27.3%)患儿入院时表现为完全性右束支阻滞合并左前分支阻滞(其中1例合并二度II型房室传导阻滞);④1例(4.5%)患儿入院时表现为完全性左束支阻滞;⑤1例(4.5%)患儿入院时即表现为完全性房室传导阻滞;⑥22例患儿中,有8例(36.4%)患儿最终进展为完全性房室传导阻滞,2例患儿进展为高度房室传导阻滞;⑦有8 例(100%)患儿在出现高度/完全性房室传导阻滞前为完全性右束支合并左前分支阻滞,平均潜伏期1.7年;⑧3例(13.7%)患儿出现局灶性房性心动过速(简称房速)合并非持续性室性心动过速(简称室速);2例患儿出现局灶性房速,1例患儿出现非持续性室速,1例患儿出现室速、室颤;⑨4 例(18.2%)患儿合并QTc 延长,最长QTc 分别为710ms、690ms、470ms、480ms。18例患儿通过基因及肌肉活检明确诊,1例患儿行基因检查确诊,3例患儿通过经典的三联征[1]临床确诊。所有患儿均无家族相关遗传病史。
2.3 治疗与预后17例患儿植入起搏器治疗(12例VVI,4例DDD),其中1例患儿因反复发生持续性室速,更换为ICD。1例患儿因室速、室颤病史,直接植入ICD。2例患儿因合并QTc间期延长,给予纳多洛尔治疗。随访过程中4例患儿死亡,1例死于心力衰竭,1例死于胰腺炎,1例死于感染性休克,另1例死因未知。其余患儿仍在随访中。
3 讨论
Kearns-Sayre综合征是一种罕见的线粒体异常疾病,患病率为10万分之1~3[7]。该病于1958 年首次描述,其特征是进行性外眼肌麻痹、视网膜色素变性,发病年龄在20岁之前。除了主要的临床症状外,诊断还需要至少以下一种合并症:小脑性共济失调、脑脊液蛋白>100 mg/d L 和心脏传导障碍[8]。该病尤其与各种内分泌和代谢疾病有关,例如矮小、糖尿病、甲状腺功能亢进、甲状旁腺功能减退症、醛固酮增多症等。内分泌异常在线粒体疾病中很常见,当疾病首次表现为没有特定家族史的内分泌疾病且神经肌肉功能正常时,通常会错过正确的病因诊断。在某些情况下,可能会出现更多症状,例如眼球震颤、近视、前庭障碍、近端肌无力、脑病发作和各种肌病[9-10]。突变的mtDNA 与正常分子共存(异质性),突变与正常mt DNA 的比例对临床症状的发生和严重程度都有影响。迄今为止,已在人类mt DNA 中鉴定出超过100 种不同的缺失,最常见的是长度为4 977 个碱基对,跨越第8469 和第13447位核苷酸[11]。
Kearns-Sayre综合征患者的心脏表现很常见,多达57% 的患者表现为充血性心力衰竭、晕厥或心脏骤停,是这类患者死亡的主要原因[2]。通过对该类病人进行电生理检查发现,原发性异常集中在房室结-His束-浦肯野系统,导致H-V 间期延长[3]。在心脏的病理研究中,发现Kearns-Sayre综合征患者的心内膜心肌活检样本中的mtDNA 缺失。由此推断,在Kearns-Sayre综合征患者中,mt DNA 的缺失可能不限于骨骼肌,器官的差异受累可能是由于不同组织中缺失的碱基数量不同所致[4]。这类病人的窦房结、房室结、束支及其分支可见脂肪浸润、纤维化,窦房结可出现明显变性及细胞坏死。这些病理及电生理方面的研究进一步解释了这类病人传导系统进行性阻滞的原因[12]。
同时,传导系统的普遍受累可导致心室除极及复极时间(QTc)延长,使得这类病人有发生恶性室性心律失常致猝死的可能性,这在笔者搜集的文献中也有相关报道。通常认为,室性心律失常可能是由心动过缓依赖性动作电位时程(APD)和QT 间期延长以及早期后除极(EADs)增加引起的。因此,这类病人的室性心律失常可能被心室起搏引起的心率增加所抑制,从中获益。然而,另有一部分Kearns-Sayre综合征患者在没有任何心动过缓的情况下,甚至在起搏器植入后出现室性心律失常。线粒体同肌浆网一样有保持细胞内钙浓度的作用,Kearns-Sayre综合征患者的线粒体受损可能导致细胞内钙浓度升高,导致延迟后除极(DADs)增加,增加室性心律失常的可能性,这类病人考虑植入ICD更能获益[6]。
此病患儿传导系统受累的可能性极高(几乎为100%),且与晕厥和心源性猝死高度相关,应密切随访患儿的心电图变化,进行适当的管理。在这22例患者中,36.4%的患儿最终出现完全性或高度房室传导阻滞,部分伴心室停搏,且这部分患儿在此前均为完全性右束支阻滞合并左前分支阻滞,由此有理由推断,双束支阻滞的发生可以预测传导阻滞的不可避免的迅速进展(平均潜伏期1.7年)。因此,疾病早期通常每3个月复查一次常规及动态心电图,如有心脏相关症状(如晕厥、心悸、胸闷等)发生,随时复查心电图,并给予相应的治疗措施。出现双束支阻滞或二度房室传导阻滞后,建议患儿尽早植入起搏器,以防止高级别房室传导阻滞、心室停搏的发生导致的严重症状或心源性猝死。AHA/ACC/HRS和ESC指南建议预防性植入起搏器作为Kearns-Sayre综合征等神经肌肉疾病患者二度、高度或三度房室传导阻滞的I级指征,无论其是否与症状相关[13]。
现有起搏器的患者应仔细随访后续是否发生室性心律失常和晕厥等,如有发生,这类病人可能需要升级到ICD[14]。如果Kearns-Sayre综合征患者确诊时既往已有室速病史或者QTc延长等情况,考虑首选植入ICD。
综上所述,Kearns-Sayre综合征患儿起病隐匿,早期表现不典型,易漏诊、误诊。由于心脏传导系统的病变及迅速进展是该类患者猝死的首要因素,应注意密切随访心电图,在出现双束支阻滞时尽早植入心脏起搏器,并继续密切观察随访患儿有无室性心律失常及晕厥等情况发生,必要时可升级为ICD。如患者确诊时合并QTc延长或存在室性心律失常等病史,可考虑首选植入ICD,防止心脏性猝死,延长患者寿命。
