嗜麦芽糖寡氧单胞菌H002的基因组分析及对铀胁迫的转录组响应
2023-04-29唐婷李汶晋李睿冯红
唐婷 李汶晋 李睿 冯红
摘要:为探究嗜麦芽糖寡氧单胞菌H002响应金属铀冲击的分子机制及生物修复铀污染的潜力,本研究从基因组和转录组两个层面进行了分析.基因组测序结果表明, H002基因组大小为5 099 056 bp, 预测编码4780个蛋白; 与其他6个近缘菌株相比, H002拥有344个独特的编码基因, 涉及膜转运、细胞运动和分泌等功能. 转录组分析显示, 在铀胁迫的早期(1 h), 差异基因主要富集于细胞运动、分泌、蛋白质和肽聚糖合成等KEGG途径. 部分分泌通道相关基因的表达下调,可降低细胞对铀离子的摄取, 进而减轻铀离子对细胞的毒性. 在铀胁迫的后期(2 h和4 h), 参与铀生物矿化的磷酸酶相关基因和能够提供还原电子的细胞色素c基因的表达上调, 能以主动方式降低铀的细胞毒性.
关键词:嗜麦芽糖寡氧单胞菌; 铀; 基因组; 转录组学; 生物修复
收稿日期: 2022-12-15
基金项目: 四川省辐射诱变技术育种平台项目(2021YFYZ0011)
作者简介: 唐婷(1997-), 女, 四川南充人, 硕士研究生, 研究方向为微生物学. E-mail: tangting@ stu.scu.edu.cn
通讯作者: 冯红. E-mail: hfeng@scu.edu.cn
Genomic analysis and transcriptomic response to
uranium stress of Stenotrophomonas maltophilia H002
TANG Ting, LI Wen-Jin, LI Rui, FENG Hong
(Sichuan Key Laboratory of Molecular Biology and Biotechnology,
College of Life Sciences, Sichuan University, Chengdu 610065, China)
To explore the molecular mechanism of Stenotrophomonas maltophilia H002 in response to metal uranium impact and the potential of bioremediation of uranium pollution, genome and transcriptome analyses were performed in this study. It was shown that the H002 genome consists of 5 099 056 bp to encode 4780 proteins. Compared with the 6 homologous strains, H002 encodes 344 unique proteins, mainly involved in membrane transport, cell motility and secretion. Transcriptome analysis revealed that in the early stage (1 h)of uranium stress, the differentially expressed genes were mainly enriched in KEGG pathways such as cell mobility and secretion, the biosynthesis of protein and peptidoglycan. The downregulation of partial secretion channel-related genes may reduce the absorption of uranium ions, hence decreasing cellular toxicity. In the later stage (2 h and 4 h)of uranium stress, upregulation of the genes encoding phosphatases involved in uranium mineralization and the cytochrome c proteins, which can provide the reducing electronics, may help reduce the toxicity to the cells.
Stenotrophomonas maltophilia; Uranium; Genome; Transcriptome; Bioremediation
1 引 言
隨着工业化的快速发展, 在带来巨大经济效益的同时, 也面临着化石能源的日益枯竭和严重的环境污染. 因此寻找储备充足而且污染小的能源迫在眉睫, 核能就是其中的一种选择. 核能是由原子核经核反应后产生的能量, 其中重金属铀的裂变反应是目前生产核能的主要方式. 但是在铀矿的开采、冶炼以及使用过程中, 不可避免的会造成一些放射性污染[1]. 这些污染物在衰变的过程中会产生放射线, 对人类和动植物都具有极大的损害. 因此, 铀放射性污染的治理也成了一个需要攻克的难题[2]. 目前铀污染的治理主要有物理、化学和生物方法, 其中物理方法主要包括焚烧、蒸馏、蒸发及倾倒等; 化学方法主要有化学沉淀、湿式氧化、酸性消化等, 这两种方法不仅需要较高的成本, 还可能带来环境污染[3], 而生物修复因其成本低及生态友好引起了人们的重视[4].
生物修复的机制主要包括细胞内积累、生物矿化、细胞表面吸附和生物还原, 其中大多数的研究集中在生物矿化和生物还原这两个方面[5]. U(VI)的生物矿化是指U(VI)与微生物相关的配体如磷酸盐、碳酸盐或氢氧化物沉淀; 铀的生物还原是指微生物可以通过还原作用将高毒性的U(VI)还原成难溶且低毒性的U(IV), 从而达到治理铀污染的作用. 例如沙雷氏菌属可以在磷酸酶的介导下将U(VI)沉淀为胞外磷酸铀矿(HUO2PO4)[6]; 硫还原菌可以通过毛状蛋白丝或菌毛还原U(VI)[7]; 奥奈达希瓦氏菌也可通过还原作用将高毒性的U(VI)还原成难溶且低毒的U(IV)[8]. 许多微生物被用于处理铀污染的废水, 但常见的微生物对辐射很敏感, 放射性核素铀的放射毒性将会影响其生存及某些特定功能, 从而限制它们在治理铀污染中的应用. 目前的研究表明, 嗜麦芽糖寡氧单胞菌耐受高水平的有毒重金属, 诸如Cd、Pb、Co、Zn、Hg、Ag和铀酰等, 并且还能在50 mmol/L的亚硒酸盐和25 mmol/L的亚碲酸盐条件下生长, 并将其分别还原为单质硒粒Se0和晶体碲Te0[9]. 因此, 嗜麦芽糖寡氧单胞菌在铀以及其它重金属离子的治理方面具有一定的潜力.
随着测序技术的快速发展, 利用组学的方法解析分子机制已成为重要的研究手段. 转录组测序可以了解微生物在去除重金属和适应环境的过程中, 胞内发生的一系列复杂的生理生化反应分子机制[10]. 分子机制的解析有助于理解细菌如何与环境中的放射性核素相互作用, 同时可以用来改进生物修复技术[11]. 本实验室前期采用150 mg/L的U(VI)进行筛菌, 获得一株对重金属铀具有高度耐受能力的嗜麦芽糖寡氧单胞菌H002, 在400 mg/L的铀溶液下, 铀的去除率为60%, 换算的吸附量为240 mg/g[12]. 为了进一步探索H002对金属铀的响应机制, 本研究针对H002进行基因组测序, 通过组装获得了完整的基因组; 并通过转录组测序, 分析研究了对铀的适应机制, 为后续的菌株改造以及铀污染的治理提供理论依据.
2 材料和方法
2.1 材 料
嗜麦芽糖寡氧单胞菌H002(CGMCC 1.18418)由本实验室在60Co辐照环境中分离获得. 菌株采用TGY培养基(0.5%蛋白胨、0.3%酵母粉、0.1%葡萄糖)于30 ℃培养.
2.2 方 法
2.2.1 基因组DNA和RNA提取
挑取H002单菌落于TGY液体培养基中震荡培养至OD600约为0.8, 离心收集菌体, 参照SDS-酶裂解法[13]提取基因组DNA. 将TGY过夜培养的新鲜菌液, 按1%接种至100 mL TGY液体培养基中, 30 ℃震荡培养, 8 h后加入终浓度为400 mg/L的U(VI)溶液, 继续在相同条件下培养, 并取菌液测定OD600. 在铀处理0 h、1 h、2 h、4 h分别收取2~3 mL菌液, 经液氮速冻后-80 ℃保存, 用于提取总RNA. 总RNA按照TRIzol试剂(购自 Thermo ScientificTM)指南提取.
2.2.2 基因组测序、组装和注释
上述基因组DNA经质检达标后(DNA浓度高于100 ng/μL, 且总量大于10 μg), 委托北京诺禾致源生物科技有限公司利用PacBio RSⅡ测序平台进行建库、测序, 原始数据已上传NCBI SRA数据库(PRJNA656069). 通过SMRT Portal(v2.3.0)平台去除低质量的碱基及adapter, 获得subreads数据, 然后采用HGAP[14]基于有向无环图的一致性进行从头组装, 最后运用Pbjelly[15]软件利用PacBio长读取数据填充gap, 获得闭合完整的基因组序列.
将组装完整的基因组序列采用Rast系统(https://rast.nmpdr.org/)[16]进行在线注释; 然后将预测的基因上传Blast2GO[17], 利用内置比对程序完成序列与Nr数据库的同源比对, 结果文件经Blast2GO数据库搜寻后获得GO注释文件. 同时将基因组序列提交至在线网站(https://www. genome.jp/tools/kaas/)[18]与KEGG数据库进行比对, 利用TBtools[19]软件进行富集分析. 最后采用Circos[20]软件绘制基因组的二维可视化图谱.
2.2.3 转录组分析
将检测合格的总RNA样品委托北京诺禾致源生物科技有限公司使用Illumina PE150平台进行双端测序, 原始数据已提交NCBI SRA数据库(PRJNA656069). 经trim_galore和multiQC[21]进行数据质控, 得到clean data, 然后提交至Hisat2[22], 根据H002基因组序列建立索引, 采用默认参数运算并获得比对文件, 运用samtools[23]软件完成文件的排序及格式转换, 再利用StringTie[24]进行定量, 使用python脚本分析数据获得转录本的count文件及TPM值; 最后, 基于R语言的edgeR[25]软件包分析count数据, 计算得到log2FC、P value和FDR等数值, 获得差异基因(DEG), 筛选阙值FDR < 0.05, 且|log2FC| > 1.
2.2.4 RT-qRCR
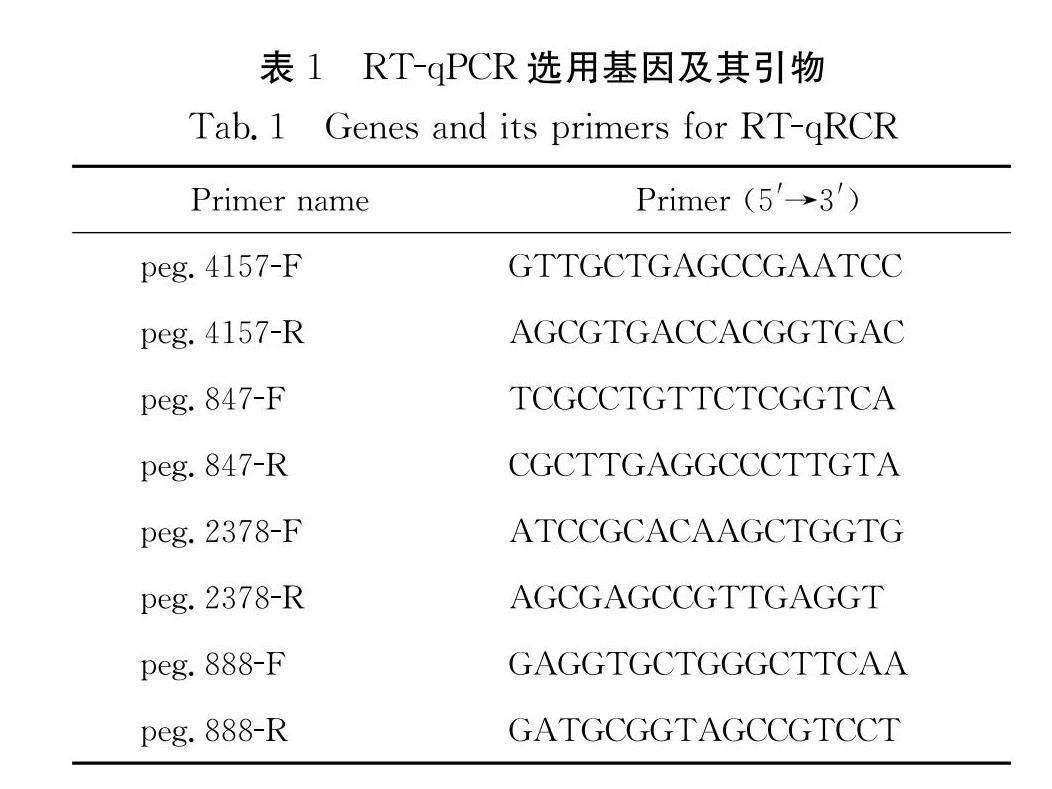
为验证RNA-seq数据的可靠性, 随机挑选了4个基因进行RT-qRCR, 分析了相关基因的相对表达量. qPCR的引物利用Primer5设计, 引物序列见表1, 并以16S rRNA基因作为内参基因.
3 结 果
3.1 基因组分析
3.1.1 S.maltophilia H002基因组组装与注释
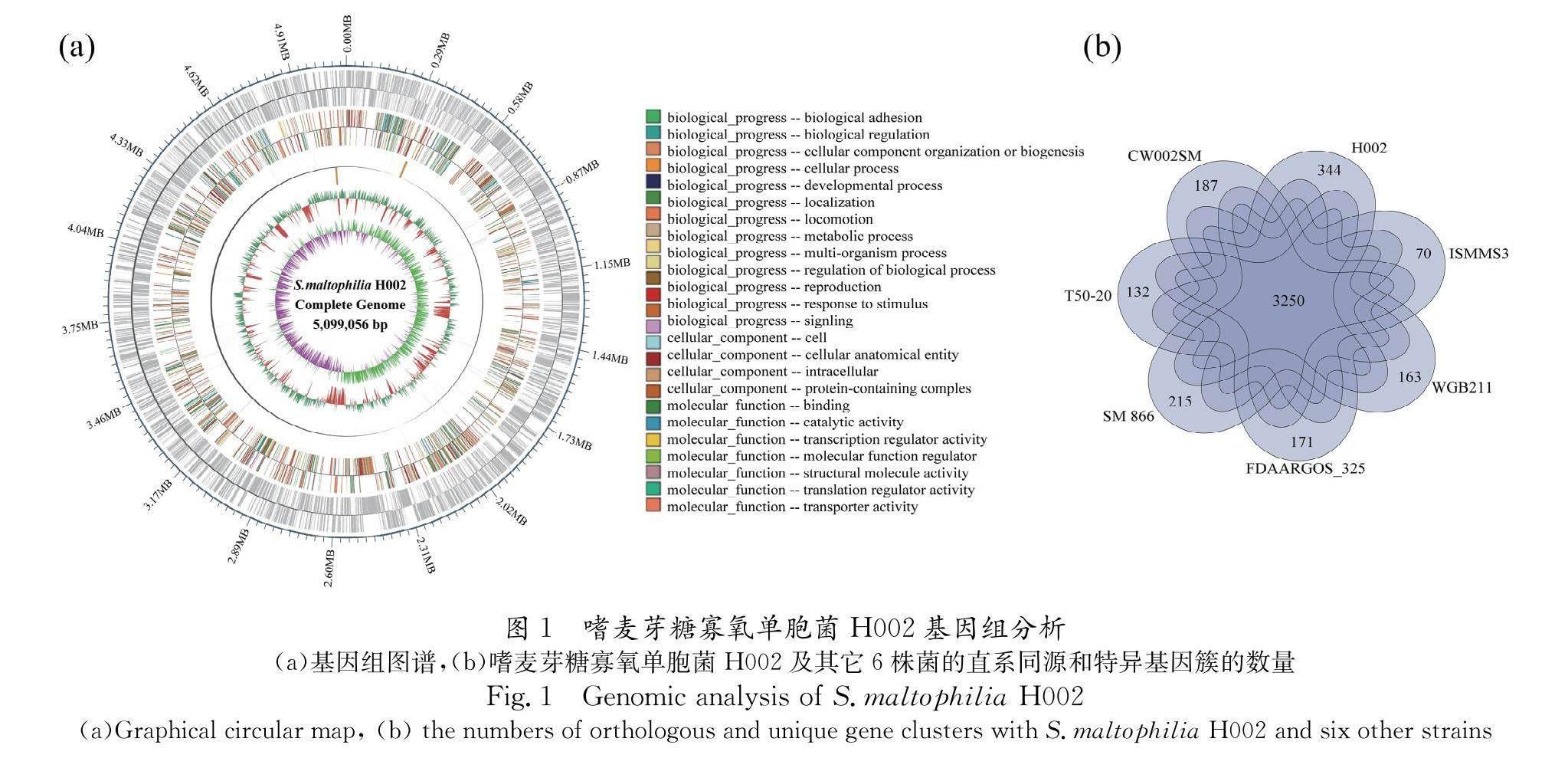
为了解嗜麦芽糖寡氧单胞菌H002的基因组特征及遗传基础, 本研究对H002菌株进行了全基因组测序. 结果表明H002菌株的基因组大小为5 099 056 bp(图1a), GC含量为66.5%. Rast预测得到4868个基因, 其中包括4780个蛋白编码基因, 13个rRNA和75个tRNA基因.
进一步分析了H002的基因功能, 将基因组提交GO数据库进行比对, 共注释3287个基因, 占总编码基因的68.76%, 主要分为三个组分, 其中生物过程(Biological process)占比48.49%, 主要参与代谢过程及细胞过程; 细胞组分(cellular component)占比24.61%, 主要参与细胞解剖实体和细胞内组分; 分子功能(molecular function)占比66.05%, 其中催化活性及结合相关的基因最多(图1a). 利用KEGG数据库进行代谢通路富集, 共注释了1942个基因, 基因分布较多的依次为次生代谢产物生物合成(Biosynthesis of secondary metabolites)、抗生素生物合成(Biosynthesis of antibiotics)、不同环境中的微生物代谢(Microbial metabolism in diverse environments)和双组分系统(Two-component system).
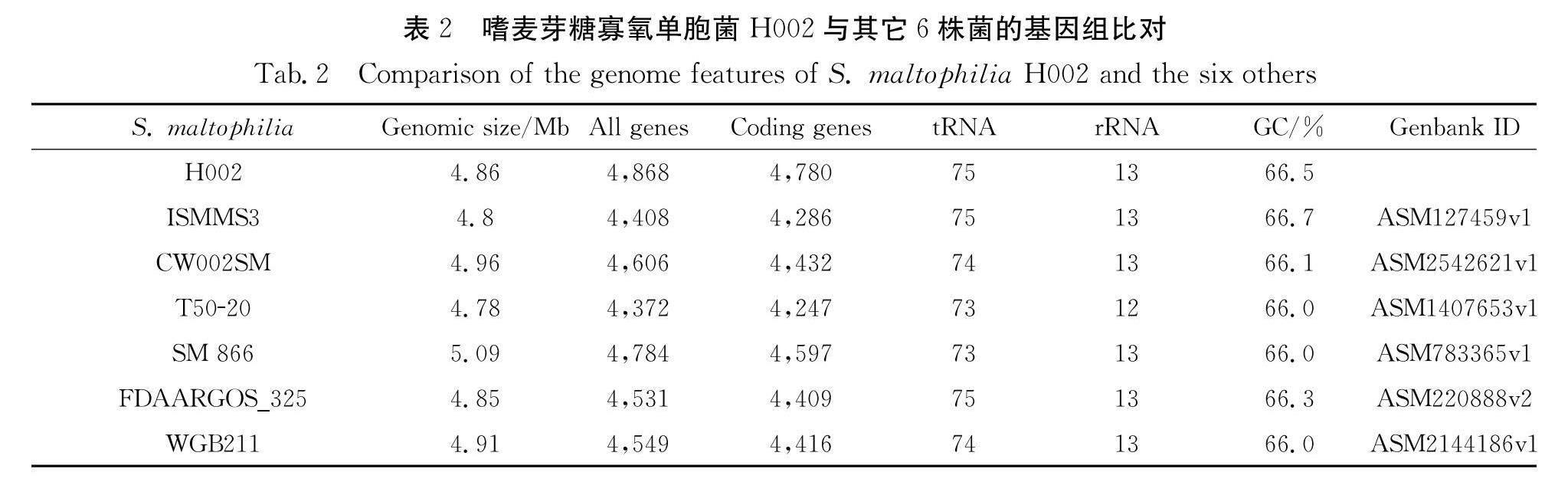
3.1.2 比较基因组分析
将H002基因组与数据库中的其它嗜麦芽糖寡氧单胞菌菌株的基因组进行了比较, 各基因组的基本特征见表2. 它们的基因组大小和GC含量均非常相似, 其中H002预测编码基因最多, 编码基因4780个. 比较基因组分析表明, H002和其它6株嗜麦芽糖寡氧单胞菌共有3250个核心蛋白(图1b), 各菌株存在的特有蛋白数分别为344个H002, 215个SM 866, 187个CW002SM, 171个FDAARGOS_325, 163个WGB211, 132个T50-20, 70个ISMMS3. 其中H002的特有基因最多, 包括310个基因编码未知蛋白; 另外 34个已知功能基因, 主要编码膜蛋白、Ⅲ型限制-修饰系统以及分泌相关的蛋白. 这些特有的基因可能与不同菌株的生境适应, 如铀的耐受相关.
3.2 对铀胁迫的转录组响应
3.2.1 铀冲击对S. maltophilia H002细胞生长的影响
为探究H002菌株在铀冲击下的响应机制, 首先测量铀冲击下细菌的生长曲线, 并在细胞生长的对数期, 即8 h加入400 mg/L的铀. 通过测定不同时间的OD600值, 用于监测细胞的生长情况(图2a). 结果表明在加入铀以后, 细胞密度增长缓慢, 说明细胞生长受到抑制; 随着时间的推移, 细胞逐步恢复生长, 细胞密度在处理4 h后, 基本达到对照水平. 说明H002细胞在铀冲击下, 经过短暂的适应, 可能通过调整细胞的代谢, 迅速恢复生长.
3.2.2 S. maltophilia H002对铀胁迫的转录响应
为了明确H002菌株响应铀胁迫的分子或代谢过程, 分别在铀处理后的不同时间点取样, 进行了比较转录组分析. 结果显示(图2b), 在铀处理后1 h与0 h相比, 发现近千个差异表达基因, 其中488个基因上调, 460个基因下调, 说明H002细胞对铀胁迫作出了快速的响应. 随着时间的延长, 处理后2 h和4h的差异基因数目继续增加, 特别是上调的基因, 在4 h达到1743个. 说明细胞通过调节基因表达, 逐渐适应胁迫环境, 细胞恢复正常生长.
为了更直观的展现铀冲击后不同恢复生长阶段的差异基因表达模式, 将3033个差异基因的表达水平(TPM值)经过归一化处理, 绘制了表达量热图(图2c). 结果表明, H002菌株经铀冲击后, 基因表达的响应大概可区分为三种表达模式. 第一类在处理初期(1 h)表达量持续上升, 主要包括双组份信号转导及代谢分泌过程的基因. 第二类为初期(1 h)表达量下降, 后期(2 h和4 h)表达量上升. 这类基因主要参与调控细胞的运动、信号转导以及DNA的复制及修复. 第三类为处理初期(1 h)表达量基本不变, 后期(2 h和4 h)表达量上调的基因, 主要涉及信号传递及DNA的复制和修复.
3.2.3 S. maltophilia H002对铀冲击初期的差异表达基因的分析
上述结果表明铀处理的早期(1 h), 细菌细胞受到铀冲击, 导致出现近千个差异表达基因. 为了进一步了解铀胁迫早期胞内的调控机制, 将铀处理1 h与0 h的差异基因进行了KEGG富集分析(图3a). 其中, 上调基因主要涉及核糖体和生物抗性及肽聚糖合成过程. 核糖体作为蛋白质翻译的机器, 在蛋白质的合成过程中必不可少. H002菌株有70个核糖体相关基因(图3b), 在铀胁迫的初期大多数表达上调, 但是随着处理时间的增长基因表达量不断下降, 说明前期细菌通过增加蛋白质的合成, 响应铀的刺激. CAMPs是宿主抵御微生物感染和微生物产物的天然防御系统的重要组成部分, 而细菌已经发展出了许多对抗CAMPs的机制. 这些抗性机制包括通过阳离子分子取代细胞表面的阴离子成分降低对CAMPs的亲和力, 膜射流泵以及肽酶的产生[26]. 此前有报道称smeDEF基因的表达能够促进嗜麦芽糖寡氧单胞菌的固有耐药性[27]. KEGG富集在CAMP抗性通道的RND型外排泵基因smeDEF(peg.4155, peg.4156, peg.4157)在铀处理1 h后其表达量急剧上升, 表达水平提高了至少4倍以上, 后续也维持高水平表达. 说明外排泵基因能够迅速响应铀的刺激, 可能具有解毒功能. 肽聚糖是细菌细胞壁的主要组成之一, 对于维持细胞形态及细胞存活是必需的, 说明细胞可能通过增强细胞壁从而抵御铀离子进入细胞.
膜分泌通道作为细菌与外界环境沟通的重要枢纽, 在细菌的应激响应中具有重要作用. 分析发现H002中具有223个膜相关蛋白, 在铀处理的初期, 有64个基因表达上调, 主要注释为RND型蛋白以及其它的外膜转运蛋白, 如金属离子转运蛋白. 金属离子外排是细菌常用的一种解毒策略, RND-HME(Resistance-nodulation-division heavy metal efflux)被认为是细菌潜在的铀代谢策略[28]. 基于H002的38个RND型相关基因绘制了表达量热图(图3c), 铀处理初期(1 h), 包括上述三个smeDEF基因在内共有16个基因上调, 其中包含注释为钴/锌/镉等重金属的外排蛋白、膜融合蛋白以及参与多药外排的转运蛋白. 但是, Na+和H+转运蛋白的表达量并未发生变化, 说明一价阳离子通道可能不参与初期的铀调控反应.
下调的基因主要参与调控细胞的分泌及运动. 其中T6SS作为膜内一个完整的分泌体系, 主要以接触依赖的方式将毒性效应物质传递给其他细胞. 有研究发现鲍曼不动杆菌对一些抗生素的耐药机制与T6SS系统有关. 例如耐四环素抗性质粒的存在会沉默T6SS的功能, 提高细菌的耐药性[29]. 基于H002细菌的14个T6SS相关基因绘制了表达量热图(图3d), 在铀冲击的早期, 几乎所有的T6SS基因均表达下调, 说明H002经铀处理后, 再短时间内可能通过调低T6SS系统的表达, 从而提高细菌的抵抗力. 细菌在面临不利环境时, 可以通过运动行为实现“避害”, 同样环境胁迫也能诱发细菌的运动发生改变甚至抑制细菌的运动. 再铀处理后初期, 大量细菌的鞭毛及纤毛相关基因的表达量下调, 说明铀对细菌的运动造成了损伤(图4b和4c).
3.2.4 S. maltophilia H002对铀冲击后期的差异表达基因的分析
为理解铀处理后期的细胞调控机制, 着重分析了处理后4 h与0 h的差异表达基因. 上调基因主要涉及细胞的膜转运、分泌及代谢, 其中RND外排基因的表达水平均有显著提高(图3c); 此外, 66个ABC转运相关基因中有54个基因的表达也上调. 说明随着铀刺激时间的增长, 细胞的防御机制全面开启. 一般来说, 细胞色素蛋白在还原过程中是必不可少的, 通过以细胞色素c作为电子载体可将电子传递给U(VI), 从而实现铀的生物还原[30]. 分析结果显示细胞色素c相关基因(peg.3260、peg.3262、peg.4325、peg.4326)的表达量上调. 为了感知和响应环境变化, 细菌进化出不同的调节系统, 包括参与对金属离子响应的双组分系统(Two-component system)[31]. 与0 h相比, 4 h时许多双组份系统的表达上调, 说明一些双组分系统能够感应金属离子, 增强下游防御基因的表达, 从而抵御铀离子的胁迫.
微生物分泌的磷酸酶可以水解溶液中的有机磷化合物产生无机磷酸盐, 后者与铀反应形成稳定的磷酸铀酰沉淀, 证明磷酸酶基因在铀的解毒过程中发挥着重要作用[32]. 基于H002中的36个磷酸酶基因的表达量热图(图4a), 显示在早期(1 h), 大多数磷酸酶基因表达变化不显著; 当细胞适应铀胁迫后, 开始持续上调表达, 4 h与1 h相比, 其中的30个基因的表达量提高1倍以上, 而膜相关磷脂磷酸酶(putative membrane-associated phospholipid phosphatase, peg.847)表达量上调最高, 达16倍. 相反, 只有3个基因的表达量下调. 这些结果说明磷酸酶基因主要在细胞适应铀胁迫环境后, 可能开始发挥解毒作用.
鞭毛是细菌表面生长的细长且弯曲的蛋白质附属丝状物, 不仅是细菌的一种运动器官, 同时还是一个分泌通道. 利用30个鞭毛组成元件的基因绘制了表达量热图(图8), 数据表明在早期阶段, 大多数下调, 在2 h时后转而持续上调表达, 其中编码鞭毛合成蛋白Flis的基因表达量上调达到10倍. 此外, 纤毛作为细菌表面延伸出来的丝状结构, 同样也是细菌的一个运动器官, 而且参与分泌及电子的传递. 据报道, 纤毛通过电子传递, 可在细胞外还原U(VI), 并且可以作为细胞的一种保护机制, 使U(IV)沉积物分散在纤毛中, 防止铀在细胞周质内过度沉淀, 从而减少对细胞的毒性作用[33]. 为此, 对38个纤毛相关基因进行了热图分析(图4c), 结果显示与0 h相比, 几乎所有的基因在受铀胁迫的早期均表达下调 (1 h); 但随着响应机制的激活, 大多数纤毛相关的基因在后期(4 h)表达转为上调. 说明在铀处理早期解毒机制尚未完全开启时, 细胞通过将铀泵出细胞以及减少铀离子的吸收从而降低铀的累积毒性, 而随着解毒相关基因的表达增强, 细菌通过运动及膜转运的增强促进铀的吸附从而完成铀的沉淀及还原.
3.2.5 RT-qPCR验证差异基因
随机选取转录组中的4个差异基因(peg.888、peg.2378、peg.847、peg.4157), 通过RT-qPCR, 分析了这4个基因在0 h、2 h、4 h的表达量, 并与转录组数据进行了比较, 结果表明这4个基因的转录组数据与qPCR数据趋势基本一致(图5), 说明基于转录组的差异基因表达水平是可靠的.
4 讨 论
铀作为一种常见的放射性元素, 存在于一些矿物中, 但含量都很低. 铀的毒性主要由其化学性质和放射性决定的, 一般来说溶解度越高, 毒性越强. 因此将高溶解度的铀转化为沉淀或可溶性低的铀矿成为铀污染处理的重要途径. 铀的生物修复就是利用一些具有还原或矿化能力的微生物来治理铀污染, 因其不同于物理和化学方法的高昂成本而广受重视. 但是, 铀对微生物细胞也具有毒性, 原因是铀离子可能与某些酶的竞争性结合或铀取代了酶的金属离子辅助因子, 从而导致酶失活, 严重时甚至导致细胞死亡[34]. 因此, 筛选具有高效生物修复潜力和高耐受性的细菌是必要的; 同时, 利用组学技术探索生物修复途径和耐受机理, 对于理解和菌株改造在环境治理的实践中同样具有重要意义.
本实验室前期分离得到了一株具有较高浓度铀吸附能力和耐受的嗜麦芽糖寡氧单胞菌H002菌株, 具有开发用于铀污染治理的潜力. 因此, 为了进一步了解H002菌株遗传基础以及耐受和吸附铀的分子机理, 本研究利用三代DNA测序并结合组学分析技术, 测序获得了H002的基因组, 并通过比较基因组分析, 发现H002菌株具有一些菌株特有的基因, 如膜蛋白和分泌通道相关的蛋白等, 这些基因可能与其耐铀机制相关.
其次, 利用RNA测序分析研究了H002菌株对铀冲击的转录组应答. 生长曲线结果表明, 在铀处理后的0~4 h内, 细胞生长速度虽有减缓, 但整体差异较小, 仍呈上升趋势. 重要的是H002能够在较短时间内适应铀胁迫, 很快恢复并维持细胞自身的生长. 在铀冲击后的4个时间点总共涉及3033个差异基因, 占总基因的63%, 主要涉及信号转导以及细胞代谢等相关的基因, 说明这些基因可以通过调节表达水平, 适应并降低铀胁迫的细胞毒性, 在铀处理4 h后细胞恢复正常生长. 值得注意的是, 在铀胁迫处理后的不同阶段, 差异表达基因类别是不同的, 说明不同的基因在响应铀胁迫发挥了不同的功能. 在铀处理后早期(1 h), 核糖体相关基因和细菌抗性相关基因及肽聚糖合成基因上调, 说明细胞的蛋白质合成量增加, 同时大量合成肽聚糖可能强化细胞壁, 有助于抵御铀离子. 此外, 膜分泌通道作为铀离子进入细胞的一道关卡, 在铀防御机制中具有重要作用, 部分膜通道相关基因如RND外排蛋白基因在铀冲击早期表达量上调, 可能降低铀在细胞内的积累[28]. 此外, VI型分泌通道T6SS相关基因的表达下调, 可能减少细胞摄取铀离子. 据报道T6SS可能负调控细菌的防御反应[29]. 因此, 在胁迫初期, H002细胞主要通过调节上述相关基因表达, 规避铀对细胞的毒性.
当H002细胞适应铀胁迫后, 即在2~4 h时间段, 大量的基因表达上调, 说明细菌的防御机制全面开启. 例如, 参与生物还原反应的细胞色素蛋白c相关基因在4 h表达量显著提高; 同时大量具备电子传递功能的鞭毛及纤毛合成相关基因的表达量也上调, 说明H002菌株可能利用生物还原将高毒性U(Ⅵ)还原成低毒性的U(Ⅳ), 从而达到解毒效果. 此外, 铀冲击后期磷酸酶相关基因的表达量得到显著的增强, 这些磷酸酶催化形成的磷酸基团很有可能参与了S. maltophilia H002与铀之间的相互作用, 使U(Ⅵ)转化为磷酸铀酰化合物沉淀, 从而避免铀对细胞的毒性.因此, 在铀胁迫后期, H002可能以主动的方式降低铀的生物有效性, 从而降低对细胞的毒性.
综上所述, S. maltophilia H002具备健全的膜通道和分泌系统、磷酸酶编码基因、电子传递系统等遗传基础, 可以通过生物还原以及生物矿化等机制将可溶性的铀污染转化为不溶性的铀, 实现铀污染的生物修复, 这与硫酸盐还原菌的修复过程相似[35]. 然而, 铀污染经常伴随Cd、Cu、Zn等重金属的共同污染, 这些重金属可能威胁硫酸盐还原菌的生长[36]. 而S. maltophilia对Cd、Cu、Zn等重金属也具有一定的耐受性[9], 因此, S. maltophilia H002在治理铀以及其他重金属污染等方面具有一定优势和开发潜力.
5 结 论
本研究利用转录组学分析了S. maltophilia H002耐受重金属铀离子的分子机制, 揭示了在铀冲击的早期阶段(1 h), H002细胞通过上调蛋白质和细胞壁的合成, 以及膜通道, 可能降低细胞吸收铀离子, 规避铀离子对细胞的损伤; 当细胞适应胁迫后, 即在2 h和4 h能够启动包括双组分系统、RND外排蛋白、磷酸酶、细胞色素c等基因的表达, 通过感应铀离子胁迫信号及传递、生物矿化和还原作用等机制主动降低铀的细胞毒性.
参考文献:
[1]Wufuer R, Wei Y, Lin Q, et al. Uranium bioreduction and biomineralization [J]. Adv Appl Microbiol, 2017, 101: 137.
[2]Wang J S, Hu X J, Liu Y G, et al. Biosorption of uranium (Ⅵ)by immobilized Aspergillus fumigatus beads [J]. J Environ Radioact, 2010, 101: 504.
[3]Natarajan V, Karunanidhi M, Raja B. A critical review on radioactive waste management through biological techniques [J]. Environ Sci Pollut Res Int, 2020, 27: 29812.
[4]Turick C E, Knox A S, Leverette C L, et al. In situ uranium stabilization by microbial metabolites [J]. J Environ Radioact, 2007, 99: 890.
[5]Merroun M L, Selenska-Pobell S. Bacterial interactions with uranium: an environmental perspective [J]. J Contam Hydrol, 2008, 102: 285.
[6]Macaskie L E, Bonthrone K M, Rouch D A. Phosphatase-mediated heavy metal accumulation by a Citrobacter sp. and related enterobacteria [J]. FEMS Microbiol Lett, 1994, 121: 141.
[7]Cologgi D L, Lampa-Pastirk S, Speers A M, et al. Extracellular reduction of uranium via Geobacter conductive pili as a protective cellular mechanism [J]. Proc Natl Acad Sci USA, 2011, 108: 15248.
[8]Wall J D, Krumholz L R. Uranium reduction [J]. Annu Rev Microbiol, 2006, 60: 149.
[9]Pages D, Rose J, Conrod S, et al. Heavy metal tolerance in Stenotrophomonas maltophilia [J].PLoS One, 2017, 3: e1539.
[10]Lu M, Jiao S, Gao E, et al. Transcriptome response to heavy metals in Sinorhizobium meliloti CCNWSX0020 reveals new metal resistance determinants that also promote bioremediation by Medicago lupulina in metal-contaminated Soil [J]. Appl Environ Microbiol, 2017, 83: e01244.
[11]Rogiers T, Houdt R V, Williamson A, et al. Molecular mechanisms underlying bacterial uranium resistance [J]. Front Microbiol, 2022, 13: 822197.
[12]陈天宇, 孙敏, 冯红. 嗜麦芽糖寡养单胞菌H002对铀的生物吸附 [J]. 四川大学学报: 自然科学版, 2021, 58: 145.
[13]乔建军, 杜连祥. 一种快速有效的枯草芽孢杆菌染色体的提取方法 [J]. 生物技术, 2001, 11: 38.
[14]Chin C S, Alexander D H, Marks P, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data [J]. Nat Methods, 2013, 10: 563.
[15]English A C, Richards S, Han Y, et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology [J]. PLoS One, 2012, 7: e47768.
[16]Overbeek R, Olson R, Pusch G D, et al. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST)[J]. Nucleic Acids Res, 2014, 42: 206.
[17]Conesa A, G?tz S, García-Gómez J M, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research [J]. Bioinformatics, 2005, 21: 3674.
[18]Moriya Y, Itoh M, Okuda S, et al. KAAS: an automatic genome annotation and pathway reconstruction server [J]. Nucleic Acids Res, 2007, 35: 182.
[19]Chen C, Chen H, Zhang Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data [J]. Mol Plant, 2020, 13: 1194.
[20]Krzywinski M, Schein J, Birol I, et al. Circos: an information aesthetic for comparative genomics [J]. Genome Res, 2009; 19: 1639.
[21]Ewels P, Magnusson M, Lundin S, et al. MultiQC: summarize analysis results for multiple tools and samples in a single report [J]. Bioinformatics, 2016, 32: 3047.
[22]Kim D, Langmead B, Salzberg S L. HISAT: a fast spliced aligner with low memory requirements [J]. Nat Methods, 2015, 12: 357.
[23]Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools [J]. Bioinformatics, 2009, 25: 2078.
[24]Pertea M, Kim D, Pertea G M, et al. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown [J]. Nat Protoc, 2016, 11: 1650.
[25]Robinson M D, McCarthy D J, Smyth G K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data [J]. Bioinformatics, 2010, 26: 139.
[26]Tzeng Y L, Ambrose K D, Zughaier S, et al. Cationic antimicrobial peptide resistance in Neisseria meningitidis [J]. J Bacteriol, 2005, 187: 5387.
[27]Zhang L, Li X Z, Poole K. SmeDEF multidrug efflux pump contributes to intrinsic multidrug resistance in Stenotrophomonas maltophilia [J]. Antimicrob Agents Chemother, 2001, 45: 3497.
[28]Sutcliffe B, Chariton A A, Harford A J, et al. Insights from the genomes of microbes thriving in uranium-enriched sediments [J]. Microb Ecol, 2018, 75: 970.
[29]Weber B S, Ly P M, Irwin J N, et al. A multidrug resistance plasmid contains the molecular switch for type Ⅵ secretion in Acinetobacter baumannii [J]. Proc Natl Acad Sci USA, 2015, 112: 9442.
[30]Lovley D R, Ueki T, Zhang T, et al. Geobacter: the microbe electric′s physiology, ecology, and practical applications [J]. Adv Microb Physiol, 2011, 59: 1.
[31]Chang C, Stewart R C. The two-component system. Regulation of diverse signaling pathways in prokaryotes and eukaryotes [J]. Plant Physiol, 1998, 117: 723.
[32]Beazley M J, Martinez R J, Sobecky P A, et al. Uranium biomineralization as a result of bacterial phosphatase activity: insights from bacterial isolates from a contaminated subsurface [J]. Environ Sci Technol, 2007, 41: 5701.
[33]You W, Peng W, Tian Z, et al. Uranium bioremediation with U(Ⅵ)-reducing bacteria [J]. Sci Total Environ, 2021, 798: 149107.
[34]Burbank K A, Walker R A, Peyton B M. A molecular level mechanism for uranium (Ⅵ)toxicity through Ca2+displacement in pyrroloquinoline quinone-dependent bacterial dehydrogenase [J]. J Inorg Biochem, 2015, 149: 59.
[35]Li X. Krumholz L R. Thioredoxin is involved in U(Ⅵ)and Cr(Ⅵ)reduction in Desulfovibrio desulfuricans G20 [J]. J Bacteriol, 2009, 191: 4924.
[36]Zheng J Y, Kai X T, Ai L T, et al. Influence of environmental factors on reductive bioprecipitation of uranium by sulfate reducing bacteria [J]. Int Biodeter Biodegr, 2007, 60: 258.
引用本文格式:
中 文: 唐婷, 李汶晋, 李睿, 等. 嗜麦芽糖寡氧单胞菌H002的基因组分析及对铀胁迫的转录组响应[J]. 四川大学学报: 自然科学版, 2023, 60: 066003.
英 文: Tang T, Li W J, Li R, et al. Genomic analysis and transcriptomic response to uranium stress of Stenotrophomonas maltophilia H002 [J]. J Sichuan Univ: Nat Sci Ed, 2023, 60: 066003.
