燕麦多肽数据库及降血糖活性的虚拟筛选
2023-04-29许丽佳李正文董宏波曹力心杜文义
许丽佳 李正文 董宏波 曹力心 杜文义
摘要:为了深入研究燕麦多肽中可能发挥降血糖功能的活性多肽分子,本文首先从文献中调研了从燕麦中提取鉴定得到的多肽,构建了对应的燕麦多肽数据库,并基于DPP4蛋白对多肽数据库进行了虚拟筛选.随后,针对筛选获得的6个多肽分别进行了100 ns的分子动力学模拟.从模拟之后稳定结合的构象分析了不同多肽分子与DPP4的相互作用信息,并分别计算了不同多肽分子与DPP4的结合自由能.结果表明,从燕麦多肽数据库中筛选得到的多肽可以与DPP4蛋白稳定结合,其中2个多肽分子与DPP4的亲和力相对较强.本文得到的多肽分子可以作为后续DPP4抑制剂设计和改造的先导分子,燕麦多肽数据库也可用于研究燕麦的其他生物学功能.
关键词:燕麦多肽;多肽数据库;虚拟筛选;分子动力学模拟;糖尿病
收稿日期: 2023-02-13
基金项目: 农业农村部杂粮加工重点实验室开放课题(2021CC006);成都大学人才培养质量和教学改革项目(cdjgb2022146)
作者简介: 许丽佳(1986-),女,四川成都人,讲师, 研究方向为天然药物的活性成分及作用机制. E-mail: xulijia@cdu.edu.cn
通讯作者: 杜文义.E-mail: modekji@modekeji.cn
Study on the oat peptide database and virtual
screening of hypoglycemic activity
XU Li-Jia1, LI Zheng-Wen1, DONG Hong-Bo1, CAO Li-Xin1, DU Wen-Yi2
(1. School of pharmacy, Chengdu University, Chengdu 610106, China;
2. Chengdu Like Technology Co., Ltd., Chengdu 610106, China)
To further study the active peptide molecules in oats that may exert a hypoglycemic function, it was first investigated the oat peptides extracted and identified from the literature, constructed the corresponding oat peptide database, and screened the peptide database based on DPP4 proteins. Subsequently, 100 ns of molecular dynamics simulations were performed for each of the six peptides obtained through the screening. The interaction information between different peptide molecules and DPP4 was analyzed from the stable binding conformations after simulation, and the binding free energy of different peptide molecules and DPP4 was calculated respectively. The results show that the peptide molecules obtained from the virtual screening can all stably bind to the DPP4 protein, and two of the peptide molecules have relatively strong affinity for DPP4. The peptide molecules obtained here can serve as lead molecules for the subsequent design and modification of DPP4 inhibitors, and the oat peptide database can also be used to study other biological functions of oat.
Oat peptides; Peptie database; Virtual screening; Molecular dynamics simulation; Diabetes
1 引 言
燕麦(Avena sativa L.)又被称为雀麦和野麦子,属于禾本科,燕麦族,燕麦属,是世界范围内广泛种植的作物,分布于五大洲42个国家,主要集中在北半球的温带地区.燕麦又可以分为带稃型和裸粒型,我国种植的燕麦以裸粒型为主,主要分布在河北、内蒙古、陕西、山西和云贵川等地区.由于燕麦口感不佳,过去经常被作为饲料或杂粮,随着人们生活水平的提升,大家对均衡膳食结构以及养生食物的关注逐渐增加,因此燕麦也渐渐走进大众的视野,成为新时代健康的研究热点.
目前研究表明,燕麦属于一种全价营养谷物,富含蛋白质、脂质、可溶性膳食纤维、维生素、矿物质和其他生物活性成分[1].燕麦中约40%~50%的成分为淀粉,其中18%~29%为直链淀粉,燕麦中淀粉的形态与燕麦的品种及生长环境相关;约15%~20%成分为蛋白质,其中包含人体所必需的8种氨基酸.相比大米和小麦,燕麦中赖氨酸和色氨酸含量较多[2,3].其中赖氨酸的含量是大米和小麦的2倍以上,但缺乏半胱氨酸和脯氨酸.此外燕麦中球蛋白的占蛋白总量的55%左右,其他蛋白的含量相对较少.燕麦中膳食纤维的含量在12%~15%左右,尤其富含β-葡聚糖,燕麦中80%的β-葡聚糖均为可溶性膳食纤维[4],有助于降低血液中胆固醇和血糖的含量,可以预防心血管疾病、肥胖等疾病[5,6].燕麦中2%~3%的成分为矿物质,主要为磷、钾、镁和钙等.燕麦中还富含维生素B1、维生素E和泛酸等.此外燕麦中的酚类物质也具有较强的抗氧化作用[7].
鉴于燕麦的多种营养价值,已被很多专家学者建议作为稻米和小麦之后的“第三主粮”[8].目前对燕麦的研究主要集中在多糖和多肽两个方面.
燕麦多糖主要是指燕麦中的非淀粉多糖,主要集中在燕麦的胚乳和糊粉层细胞壁,主要成分为β-葡聚糖.β-葡聚糖作为一种可溶性膳食纤维,其化学结构和理化性质已被广泛研究[9-11].燕麦中的β-葡聚糖由均已的D-吡喃葡糖糖单元通过β(1-3)和(1-4)糖苷键连接成的线性多糖.研究表明,在食用β-葡聚糖后,可以对人体产生有益的影响,主要包括降低胆固醇、调节肠道菌群、抗肿瘤和免疫刺激作用等[12-14].Shen等[15]通过研究发现β-葡聚糖可以显著增加小鼠体内肠道中乳酸菌和双歧杆菌的数量,并抑制大肠杆菌的生长;Yang[16]采用体外发酵实验验证了添加燕麦β-葡聚糖可以提高肠道菌群中厚壁菌的含量;Nakashima等[17]也通过实验证明燕麦β-葡聚糖具有抗氧化和抗肿瘤的作用.综上,燕麦多糖作为一种功能性膳食纤维,具有多种生理活性,在食品工业中具有很大的应用前景.
燕麦中含有大量的蛋白质成分,这些蛋白质在体内可以被蛋白酶水解释放出多肽片段,可以发挥特定的功能多肽片段,称为生物活性肽.具体来讲,生物活性肽是指具有一定生理作用的肽类化合物,其分子量介于氨基酸和蛋白质之间,通常由几个到几十个氨基酸通过肽键连接而成,并且可以被磷酸化,糖基化和酰基化修饰[18].大多数生物活性肽以非活性状态存在于蛋白质中,当被水解释放之后,其生物活性才会被激活.目前对于燕麦中的蛋白多肽研究相对较少,尤其是燕麦多肽在降血糖方面的作用.研究表明,燕麦蛋白中存在大量的生物活性肽分子,因此本项目拟采用计算机辅助药物设计方法研究燕麦中的活性多肽对糖尿病相关靶点二肽基肽酶的分子识别和抑制机理,从而为基于燕麦生物活性的新型保健食品开发提供理论依据.
二肽基肽酶是一种位于细胞表面的丝氨酸肽酶,属于膜蛋白,广泛存在于肾脏、结缔组织、胃肠道和淋巴结中.胰高血糖素样肽1(GLP-1)和葡糖糖依赖性胰岛素释放肽(GIP)可以诱导β细胞分化,刺激胰岛素的生成和释放,但它们的半衰期非常短,在体内会快速被DPP4降解[19].因此通过抑制DPP4来延长体内GLP-1和GIP的半衰期,是目前治疗糖尿病的重要方法[20].目前已知的DPP4抑制剂大多为多肽类化合物.另外,实验研究表明,燕麦具有较好的降血糖功效.因此,基于燕麦多肽的DPP4抑制剂研究具有一定的代表性和可行性.
2 材料与方法
2.1 燕麦多肽数据库构建
鉴于燕麦中含有大量多肽成分,为了深入研究多肽成分的功能,本文搜集了近十年的文献中的数据[21-33],整理并优化,构建得到含有140条多肽的燕麦多肽数据库.采用YASARA软件[34]的Build模块分别构建140个燕麦多肽的三维结构,并采用Gromacs 2019.6程序[35]对每个多肽分子在水溶液中进行10 ns的MD模拟,取MD模拟之后的结构作为优化好的多肽结构,并构建成.sdf数据库文件.多肽数据库的下载链接为:https://www.modekeji.cn/peptide_database.
2.2 虚拟筛选
从蛋白质数据库(Protein data bank, PDB)中选择2BGR结构[36],该晶体结构中含有已知的DPP4多肽抑制剂(多肽序列为MDP),删掉晶体结构中已有的多肽抑制剂分子和水分子,将该结构作为虚拟筛选的受体结构.
采用Schrodinger程序中的Virtual Screening Workflow模块对构建的燕麦多肽数据库进行虚拟筛选.导入优化好的燕麦多肽数据库文件,并将2BGR晶体结构中的多肽结合位置设置为虚拟筛选的结合位点.分别对多肽数据库中的多肽进行高通量对接筛选(High throughput virtual screening,HTVS)、标准精度筛选(Standard precision, SP)和高精度筛选(Extra precision, XP),其中HTVS筛选得到的多肽分子取排名前50%进行SP筛选,然后再取排名前20%的多肽分子进行XP筛选计算.
虚拟筛选之后的结果在空间结构上可能有不合理的原子接触,可以采用能量优化的方法对这些作用力进行释放,使其更趋于稳定结构.能量优化采用Amber14力场[37],优化过程分两步进行:先进行2000步的最陡下降法优化,再用2000步的共轭梯度法对结构进行进一步优化,将最终的结果作为后续分析的模型.
2.3 分子动力学模拟
为了进一步研究多肽与DPP4的分子识别驱动力,本文采用分子动力学(Molecular dynamics, MD)模拟对虚拟筛选获得的DPP4蛋白与多肽的复合物结构进行模拟研究.MD模拟采用Gromacs 2019.6程序[35],在恒温恒压以及周期性边界条件下进行.应用Amber14SB全原子力场[37],TIP3P水模型[38].在MD模拟过程中,所有涉及氢键采用LINCS算法[39]进行约束,积分步长为2 fs.静电相互作用采用(Particle-mesh Ewald)PME方法[40]计算.非键相互作用截断值设为10 ,每10步更新一次.采用V-rescale [41]温度耦合方法控制模拟温度为300 K,采用Parrinello-Rahman方法[42]控制压力为1 bar.首先,采用最陡下降法对两个体系进行能量最小化,以消除原子间过近的接触;然后,在300 K进行100 ps的NVT平衡模拟;最后,分别对6个不同体系分别进行100 ns的MD模拟,每10 ps保存一次构象,模拟结果可视化采用Gromacs内嵌程序和VMD完成.
2.4 结合自由能及能量分解
结合自由能计算采用gmx_MMPBSA工具[43],调用Amber18的MMPBSA模块进行计算.同时采用MMPBSA方法将结合自由能进行能量分解.能量分解的基本思想是将每个氨基酸的能量贡献近似分为分子力学计算的真空分子内能、用泊松-玻尔兹曼(Poisson-Boltzmann, PB)模型[44]计算的极性溶剂化能,以及用LCPO模型[45]计算的非极性溶剂化能,并将能量分解到氨基酸的主链和侧链原子上.其中真空分子内能可以分为极性的静电相互作用和非极性的范德华相互作用.通过能量分解,可以观察到DPP4和多肽结构中对二者的分子识别及结合所做出的能量贡献.
结合自由能及能量分解的计算采用MD模拟60~100 ns的轨迹,每隔200 ps取一次构象,共200个构象.
2.5 自由能曲面
自由能曲面(Free energy landscape,FEL)是通过研究体系在模拟过程中的自由能曲面图的极小值和分界点来表示生物学过程中发生的动力学变化的一种方法.一些主成分(Principal component,PC)可以表示重要的动力学过程,这些PC也可以用来绘制自由能曲面图.具体的计算公式:
△G(X)=-kBT·lnP(X)
式中,坐标X表示PC,kB表示玻尔兹曼常数,T表示绝对温度,P(X)表示某个特定的构象对整体PC的贡献,即构象分布的概率.
3 结果与分析
3.1 虚拟筛选结果
虚拟筛选的结果打分如表1所示.从表中可以看出,比晶体结构中的多肽抑制剂MDP打分更高的燕麦多肽有5个,其中KQGDVIA多肽的虚拟筛选打分最高.值得注意的是,在排名靠前的多肽分子结构中都有1~2个带电荷的氨基酸残基,而不带电荷的多肽分子的筛选排名靠后,因此根据虚拟筛选结果可以推测,多肽的带电性对DPP4的抑制活性可能有一定程度的影响.
为了进一步研究多肽与DPP4蛋白的亲和力及结合模式,根据虚拟筛选结果,本文选择排名前6的多肽进行后续的MD模拟研究.
3.2 MD模拟的收敛性分析
均方根偏差(Root mean square deviation, RMSD)表示某一时刻的构象与目标构象所有原子偏差的加和,是衡量体系是否稳定的重要依据.图1a给出了DPP4蛋白与多肽的六个体系氨基酸骨架原子的RMSD随时间的变化.从图中可以看出,六个体系的RMSD值均在0.2 nm附近波动,表明六个体系在模拟过程中均没有出现较大的结构变化.具体来讲,在模拟前40 ns内,体系的波动幅度相对较大,主要是由于在模拟初期,DPP4蛋白与周围水溶剂的相互作用较强,导致蛋白表面与水接触的氨基酸结构波动相对较大,经过一段时间的MD模拟,结构波动逐渐趋于稳定,体系RMSD值的波动也逐渐减小;在60 ns后六个体系的RMSD值基本稳定,DPP4-KQGDVIA、DPP4-AQIPEQVR、DPP4-KLPGF、DPP4-GEHGSDGNV、DPP4-FEELN和DPP4-MDP体系的平均值分别为(0.154±0.011)nm、(0.175±0.018)nm、(0.182±0.023)nm、(0.244±0.026)nm、(0.205±0.021)nm和(0.213±0.024)nm.
回转半径(Radius of Gyration, Rg)可以用来描述整体结构的变化情况,Rg变化越大表明体系结构变化越大.DPP4蛋白结构呈球形,在模拟过程中的Rg变化如图1b所示.从图中可以看出,六个体系的Rg值在模拟过程中均处于比较稳定的状态,平均值分别为(2.702±0.007)nm、(0.710±0.010)nm、(0.715±0.009)nm、(2.714±0.007)nm、(2.721±0.007)nm和(2.708±0.007)nm.整体来讲,六个复合物体系的整体形状在模拟过程中没有发生明显的变化.
综上所述,通过分析复合物体系在MD模拟过程中的RMSD和Rg变化,可以发现,六个体系中蛋白结构没有发生明显的变化,体系在MD模拟过程中均达到了稳定状态,因此得到的轨迹可用于后续的进一步分析.
为了分析多肽分子对蛋白结构的影响,本文对MD模拟过程中的蛋白结构性质变化进行了分析.两个体系在MD模拟过程中的溶剂可及表面积(SASA)如图2所示,从图中可以看出,六个体系的SASA值在模拟过程中没有出现比较明显的变化,均在320 nm2附近波动,具体来讲,DPP4-KQGDVIA、DPP4-AQIPEQVR、DPP4-KLPGF、DPP4-GEHGSDGNV、DPP4-FEELN和DPP4-MDP六个体系在60 ns后的SASA平均值分别为(320.608±2.925)nm2、(326.663±3.139)nm2、(325.301±3.287)nm2、(327.180±3.177)nm2、(325.469±3.874)nm2和(321.813±3.237)nm2,整体来讲,六个体系中DPP4蛋白的SASA平均值相差不大,波动幅度均在1%左右,表明体系的结构在模拟过程中没有明显的变化.
均方根涨落(Root mean square fluctuation, RMSF)可以表示蛋白质氨基酸残基的柔性大小,DPP4蛋白在结合不同多肽分子之后的氨基酸柔性分布如图3所示.从图3a中可以看出,六个体系的氨基酸柔性分布趋势基本一致,其中柔性较大的区域主要分布在F240~K250.从图3b中可以看出,该区域主要位于蛋白表面裸露的β-折叠及loop区域.另外从图3b中还可以看出,氨基酸柔性较大的区域(RMSF > 0.1)主要分布在DPP4蛋白表面的loop区,这些区域的氨基酸残基由于受到水溶剂的相互作用较强,导致氨基酸侧链的结构波动相对较大,因而其RMSF值较大.这些区域由于距离DPP4活性中心较远,因此较大的柔性也不会对多肽抑制剂的结合造成明显的影响.
3.3 多肽与蛋白的相互作用
3.3.1 氢 键
氢键是生物大分子体系中相互作用的重要形式,前面的分析结果表明,六个体系在模拟过程中均达到了相对稳定的状态,因此为了进一步分析氢键在多肽与DPP4蛋白分子识别过程中的作用,本文统计分析了MD模拟过程中,多肽与DPP4蛋白之间氢键数量的变化,如图4所示.从图中可以看出,在模拟前20 ns内,复合物中多肽与蛋白之间的氢键数量波动相对较大,这主要是由于在模拟初期多肽分子与附近的水分子相互作用较强,与DPP4的相互作用尚未达到稳定状态.在MD模拟60 ns后,六个体系中多肽与DPP4蛋白之间的氢键数量波动减小并趋于稳定.DPP4-KQGDVIA、DPP4-AQIPEQVR、DPP4-KLPGF、DPP4-GEHGSDGNV、DPP4-FEELN和DPP4-MDP六个体系中氢键数量的平均值分别为7.47、9.00、4.34、9.52、3.74和6.07,整体来讲,多肽AQIPEQVR和GEHGSDGNV与DPP4之间的氢键作用相对较强,而KLPGF和FEELN的氢键作用相对较弱.
3.3.2 结合模式
DPP4酶的底物结合位点主要可以分为三个(S1, S2和S3),其中,Ser630、Asn710和His740组成S1结合位点;Arg125、Glu205、Glu206和Tyr662组成了S2结合位点;S3结合位点则主要由Ser209、Arg358和Phe357组成.从DPP4结合位点的氨基酸组成可以看出,DPP4的底物结合位点中含有较多的带电荷的氨基酸残基(如Arg125、Glu205、Glu206、Arg358),因此在多肽抑制剂的结构中一般都存在1~2个带电荷的氨基酸,这样可以与DPP4的结合位点形成较强的静电相互作用.
为了分析多肽与DPP4蛋白之间分子识别的机制,本文对六种多肽分子与DPP4蛋白的结合模式进行了研究,结果如图5~图10所示.从图5a中可以看出,多肽KQGDVIA主要结合在DPP4蛋白的亲水性表面,多肽的N端结构可以伸入到DPP4的结合位点内部,形成了紧密的结合.从图5b和5c中可以看出,多肽分子主要结合在DPP4蛋白的S1和S2结合位点处,具体来讲,多肽KQGDVIA分别与Glu205、Glu206、Ser630和His740形成了氢键相互作用,另外还与Phe357、Tyr547、Trp629、Tyr662、Tyr666等疏水性氨基酸形成了较强的疏水作用,进一步增强了多肽与DPP4的亲和力.图5d给出了多肽分子与DPP4氨基酸的弱相互作用分析,从图中可以看出,二者的相互作用中氢键(蓝色区域)相对较多,另外还有一部分范德华相互作用(绿色区域).
图6给出了多肽33(AQIPEQVR)与DPP4的结合模式.从图6a中可以看出,多肽AQIPEQVR主要结合在DPP4蛋白的亲水性表面,多肽的N端结构可以伸入到DPP4的结合位点内部,形成了紧密的结合.从图6b和6c中可以看出,多肽分子主要结合在DPP4蛋白的S1、S2和S3结合位点处,具体来讲,多肽AQIPEQVR分别与Arg125、Glu205、Glu206、Asp302、Glu361、Arg356、Ile405、Ser552、Tyr662和Ser630形成了氢键相互作用,另外还与Phe357、Tyr547、Trp585、His740等疏水性氨基酸形成了较强的疏水作用,进一步增强了多肽与DPP4的亲和力.图6d给出了多肽分子与DPP4氨基酸的弱相互作用分析,从图中可以看出,二者的相互作用中氢键(蓝色区域)相对较多,另外还有一部分范德华相互作用(绿色区域).
图7给出了多肽77(KLPGF)与DPP4的结合模式.从图7a中可以看出,多肽KLPGF主要结合在DPP4蛋白的亲水性表面,多肽的N端结构可以伸入到DPP4的结合位点内部,形成了紧密的结合.从图7b和7c中可以看出,多肽分子主要结合在DPP4蛋白的S2结合位点处.具体来讲,多肽KLPGF分别与Glu205、Glu206和Asp663形成了氢键相互作用,另外还与Phe357、Tyr547、Tyr662、Tyr666等疏水性氨基酸形成了较强的疏水作用,进一步增强了多肽与DPP4的亲和力.图7d给出了多肽分子与DPP4氨基酸的弱相互作用分析,从图中可以看出,二者的相互作用中氢键(蓝色区域)相对较多,另外还有一部分范德华相互作用(绿色区域).
图8给出了多肽134(GEHGSDGNV)与DPP4的结合模式.从图8a中可以看出,多肽GEHGSDGNV主要结合在DPP4蛋白的亲水性表面,多肽结构平躺在DPP4蛋白的活性位点附近.从图8b和8c中可以看出,多肽分子没有与三个活性位点的氨基酸形成较强的相互作用.具体来讲,多肽GEHGSDGNV分别与Asp302、Arg358、Glu361、Glu408、Tyr547、Tyr585形成了氢键相互作用,与Glu408和Arg356形成了较强的静电相互作用,另外还与Pro359、Ile405、Trp629和His740等疏水性氨基酸形成了较强的疏水作用,进一步增强了多肽与DPP4的亲和力.图8d给出了多肽分子与DPP4氨基酸的弱相互作用分析,从图中可以看出,二者的相互作用中氢键(蓝色区域)相对较多,另外还有一部分范德华相互作用(绿色区域).
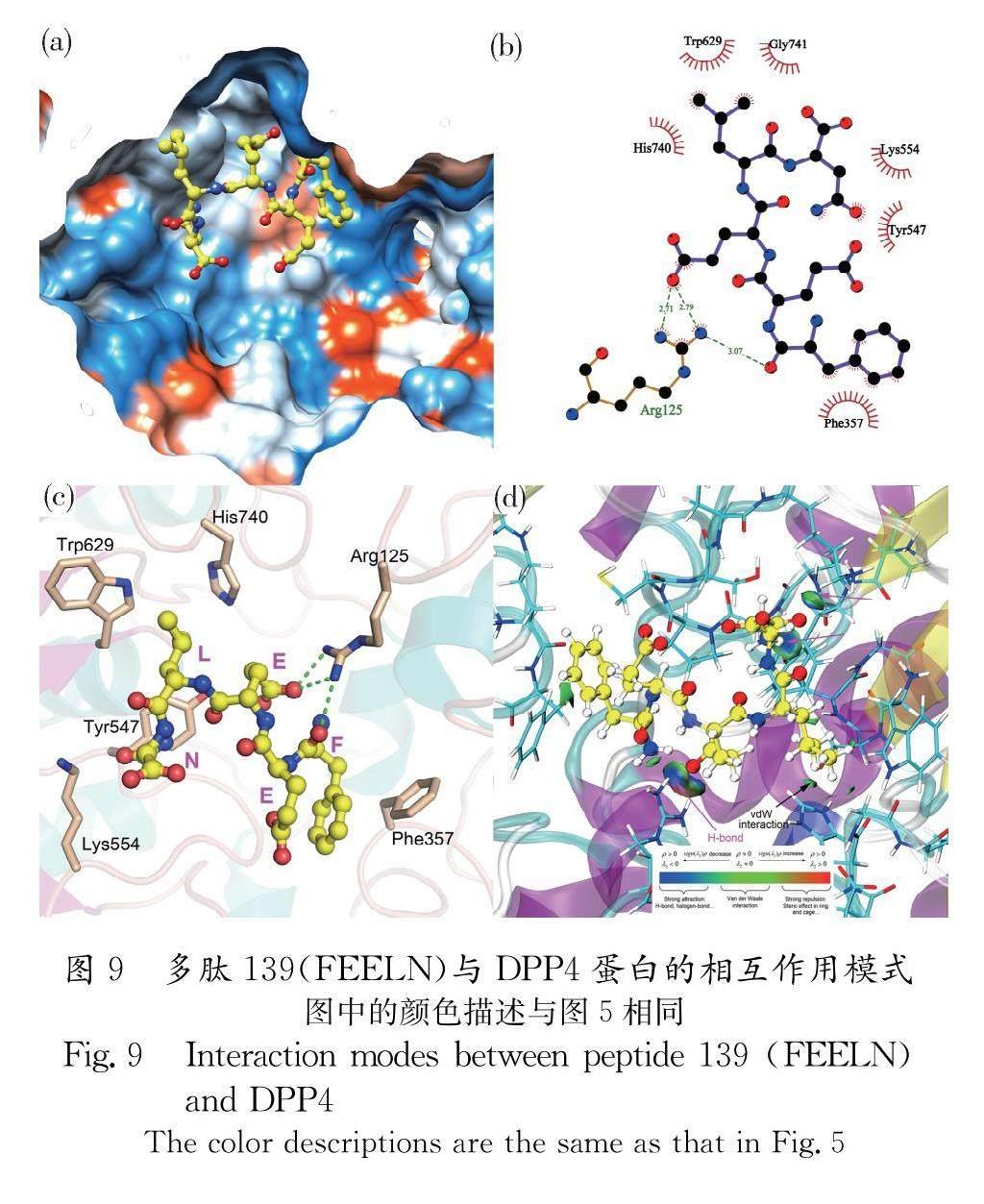
图9给出了多肽139(FEELN)与DPP4的结合模式.从图9a中可以看出,多肽FEELN主要结合在DPP4蛋白的亲水性表面.从图9b和9c中可以看出,多肽分子主要结合在DPP4蛋白的S2结合位点附近.具体来讲,多肽FEELN仅与Arg125形成了氢键相互作用,另外还与Phe357、Tyr547、Trp629、His740等疏水性氨基酸形成了较强的疏水作用,进一步增强了多肽与DPP4的亲和力.图9d给出了多肽分子与DPP4氨基酸的弱相互作用分析,从图中可以看出,二者的相互作用中氢键(蓝色区域)相对较多,另外还有一部分范德华相互作用(绿色区域).
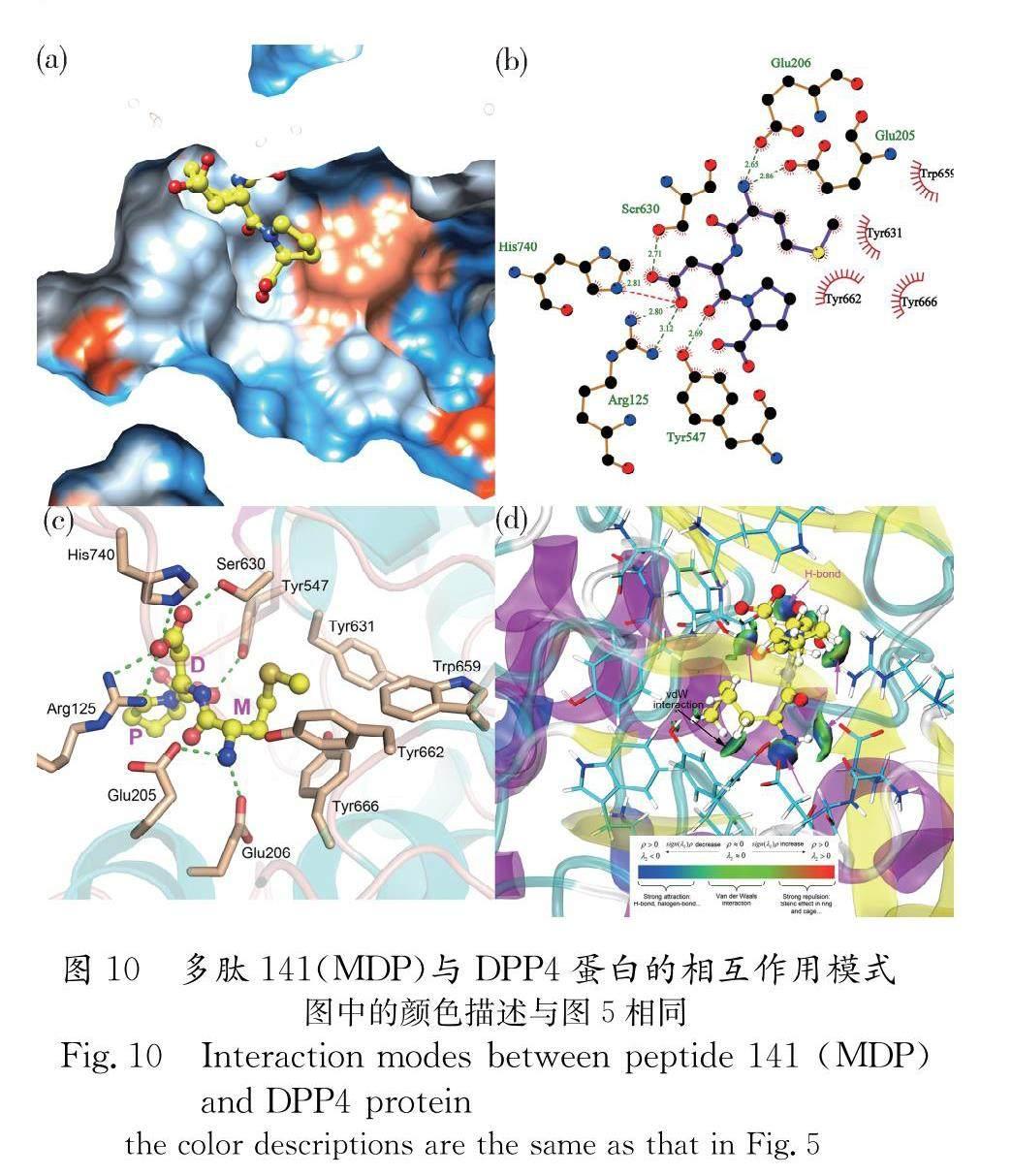
图10给出了多肽141(MDP)与DPP4的结合模式.从图10a中可以看出,多肽MDP主要结合在DPP4蛋白的亲水性表面,MDP结构的N端结构伸入到蛋白空腔内部,形成了较稳定的结合.从图10b和10c中可以看出,多肽分子主要结合在DPP4蛋白的S1和S2结合位点处.具体来讲,多肽MDP分别与Arg125、Glu205、Glu206、Tyr547、Ser630形成了氢键相互作用,与His740形成了较强的静电作用;另外还与Tyr631、Tyr662、Tyr666、Trp659等疏水性氨基酸形成了较强的疏水作用,进一步增强了多肽与DPP4的亲和力.图10d给出了多肽分子与DPP4氨基酸的弱相互作用分析,从图中可以看出,二者的相互作用中氢键(蓝色区域)相对较多,另外还有一部分范德华相互作用(绿色区域).
综上所述,在晶体结构DPP4-MDP体系中,多肽与DPP4的S1和S2位点形成了较强的相互作用.而在筛选获得的5种多肽结构中,仅有GEHGSDGNV没有明确与三个结合位点的氨基酸形成较强的氢键作用,其他4个多肽分子均与三个结合位点中的氨基酸形成了较强的氢键和静电相互作用.整体来讲,虚拟筛选获得的多肽在结合模式上,均具有潜在的抑制DPP4酶的活性.
3.3.3 自由能面图
为了进一步研究六个体系在MD模拟过程中的能量最低构象分布,本文分别分析了六个体系的自由能面图,结果如图11所示,深蓝色区域表示能量较低的构象.从图11中可以看出,六个体系中的主成分PC1和PC2变化范围并不完全相同,PC1的差别较大,其中多肽AQIPEQVR、KLPGF和MDP三个体系的PC1范围均在-4~4之间,表明这三个体系中蛋白构象变化的范围相差不大.而KQGDVIA和FEELN多肽体系的PC1范围相对较小,而GEHGSDGNV多肽体系的PC1范围相对较大.具体来讲,KQGDVIA、GEHGSDGNV和FEELN多肽体系中,自由能面图中的蓝色区域相对比较分散,蓝色区域之间有较多的绿色或红色区域,表明不同的能量最低构象之间具有较大的能垒,其构象容易落入局部能量极小势阱,而难以越过势垒达到全局能量最低的构象.在AQIPEQVR、KLPGF和MDP三个体系中,自由能面图中的蓝色区域相对比较集中,表明在模拟过程中体系达到全局能量最低构象所需要越过的势垒较小,体系比较容易达到稳定的状态.
3.3.4 结合自由能
前面从构象方面分析了不同多肽与蛋白结合的差异,而不同的结合模式会导致结合能的差异,因此本文采用MMPBSA方法分别计算了不同多肽与DPP4蛋白的结合自由能以及各个能量项的贡献,如表2所示.从表中可以看出,晶体结构中的多肽141与DPP4的结合能为-25.44 kcal/mol.在其他多肽分子中,仅有多肽AQIPEQVR和KLPGF与DPP4的结合能超过晶体结构中的多肽分子MDP,而多肽GEHGSDGNV和FEELN与DPP4蛋白的亲和力相对较弱,结合能分别为-16.59 kcal/mol 和-16.39 kcal/mol.整体来讲,多肽AQIPEQVR和KLPGF与DPP4的结合能相对较强,可以作为DPP4多肽抑制剂的先导分子.4 结 论
本文采用虚拟筛选、分子对接和MD模拟方法分别研究了六种不同燕麦多肽与DPP4的分子识别机制.采用虚拟筛选方法从燕麦多肽数据库中获得与DPP4蛋白亲和力较强的六种多肽分子,MD模拟结果表明,六种多肽可以稳定结合在DPP4蛋白的活性位点,氢键和范德华作用是多肽与DPP4分子识别的重要驱动力.其中GEHGSDGNV多肽没有与DPP4蛋白的S1/S2/S3活性位点结合,可能表明GEHGSDGNV多肽对DPP4的抑制作用较弱,其余多肽均结合在DPP4的三个活性位点处.结合自由能的计算结果表明,AQIPEQVR和KLPGF多肽与DPP4蛋白的亲和力相对较强,可以作为后续DPP4抑制剂设计和改造的先导分子.模拟结果对DPP4蛋白抑制剂的筛选和设计具有一定的指导意义.
参考文献:
[1]魏决, 罗雯. 燕麦中蛋白质的提取、纯化及氨基酸成分分析[J]. 成都大学学报: 自然科学版, 2007, 26: 283.
[2]周素梅, 申瑞玲. 燕麦的营养及其加工利用[M]. 北京: 化学工业出版社, 2009: 26.
[3]Tosh S M, Miller S S. Oats: in encyclopedia of food and health[M]. Oxford: Academic Press, 2016: 119.
[4]汪海波, 刘大川, 崔邦梓, 等. 燕麦β-葡聚糖对正常小鼠及四氧嘧啶致糖尿病小鼠血糖、血脂的调节作用研究[J]. 食品科学, 2004, 25: 172.
[5]Agostoni C, Bresson J, Fairweather-Tait S, et al. Scientific opinion on dietary reference values for carbohydrates and dietary fibre[J]. EFSA J, 2010, 8: 1462.
[6]Nazare J A, Laville M, Biliaderis C G, et al. β-Glucans in novel food ingredients for weight control[C]∥Henry C J K. Novel food ingredients for weight control. Cambridge, UK: Woodhead Publishing, 2007: 131.
[7]王凤. 燕麦面团的物性改善及其在燕麦面条中的应用[D]. 无锡: 江南大学, 2009.
[8]王洪新. 食品新资源[M]. 北京: 中国轻工业出版社, 2002: 487.
[9]Doublier J L, Wood P J. Rheological properties of aqueous solutions of (1-3)(1-4)-β-D-Glucan from oats (Avena sativa L.)[J]. Cereal Chem, 1995, 72: 335.
[10]Tosh S M, Wood P J, Wang Q,et al. Structural characteristics and rheological properties of partially hydrolyzed oat β-glucan: the effects of molecular weight and hydrolysis method[J]. Carbohydr Polym, 2004, 55: 425.
[11]申瑞玲, 董吉林, 姚惠源. 燕麦β-葡聚糖的结构研究[J]. 中国粮油学报, 2006, 21: 44.
[12]Naumann E, van Rees A B, nning G, et al. β-Glucan incorporated into a fruit drink effectively lowers serum LDL-cholesterol concentrations [J]. Am J Clin Nutr, 2006, 83: 601.
[13]Chen J Z, Huang X F. The effects of diets enriched in beta-glucans on blood lipoprotein concentrations [J]. J Clin Lipidol, 2009, 3: 154.
[14]Whitehead A, Beck E J, Tosh S, et al. Cholesterol-lowering effects of oat β-glucan: a meta-analysis of randomized controlled trials [J]. Am J Clin Nutr, 2014, 100: 1413.
[15]Shen R L, Dang X Y, Dong J L, et al. Effects of oat β-glucan and barley β-glucan on fecal characteristics, intestinal microflora and intestinal bacterial metabolites in rats [J]. J Agric Food Chem, 2012, 60: 11301.
[16]Yang J, Martinez I, Walter J, et al. In vitro characterization of the impact of selected dietary fibers on fecal microbiota composition and short chain fatty acid production [J]. Anaerobe, 2013, 23: 74.
[17]Nakashima A, Yamada K, Iwata O, et al. β-glucan in foods and its physiological functions [J]. J Nutr Sci Vitaminol, 2018, 64: 8.
[18]张开平, 苏仕林, 刘燕丽, 等. 生物活性肽功能及制备方法的研究进展[J].农产品加工, 2015, 12:61.
[19]Zeng S G, Xie H, Zeng L L, et al. Discovery of potent dipeptidyl peptidase Ⅳ inhibitors through pharmacophore hybridization and hit-to-lead optimization [J]. Bioorg Med Chem, 2013, 21: 1749.
[20]郭衔, 王翼, 龙兆丰, 等. 口服降糖药二肽基肽酶Ⅳ(DPP-4)抑制剂的多器官保护作用[J]. 药学与临床研究,2014, 22: 524.
[21]Wang F, Zhang Y, Yu T, et al. Oat globulin peptides regulate antidiabetic drug targets and glucose transporters in Caco-2 cells[J]. J Funct Foods, 2018, 42: 12.
[22]Ding L, Ma R, You H, et al. Identification and characterization ofdipeptidyl peptidase IV inhibitory peptides from wheat gluten proteins[J]. J Cereal Sci, 2022, 103: 103396.
[23]Wang F, Yu G, Zhang Y, et al. Dipeptidyl peptidase Ⅳ inhibitory peptides derived from oat (Avena sativa L.), buckwheat (Fagopyrum esculentum), and highland barley (Hordeum vulgare trifurcatum (L.)Trofim)proteins[J]. J Agric Food Chem, 2015, 63: 9543.
[24]Wang W, Liu X, Li Y, et al. Identification and characterization of dipeptidyl peptidase-IV inhibitory peptides from oat proteins[J]. Foods, 2022, 11: 1406.
[25]Montoya-Rodríguez A, Gómez-Favela M A, Reyes-Moreno C,et al. Identification of bioactive peptide sequences from amaranth (Amaranthus hypochondriacus)seed proteins and their potential role in the prevention of chronic diseases [J]. Compr Rev Food Sci Food Saf, 2015, 14: 139.
[26]Baakdah M M, Tsopmo A. Identification of peptides, metal binding and lipid peroxidation activities of HPLC fractions of hydrolyzed oat bran proteins[J]. J Food Sci Technol, 2016, 53: 3593.
[27]Ramirez F L. Development and characterization of peptides withantidiabetic activities from oat protein[D]. Canada: University of Alberta,2021.
[28]Comino I, Bernardo D, Bancel E, et al. Identification and molecular characterization of oat peptides implicated on coeliac immune response[J]. Food Nutr Res, 2016, 60: 30324.
[29]Ma S, Zhang M, Bao X, et al. Preparation of antioxidant peptides from oat globulin[J]. CyTA J Food, 2020, 18: 108.
[30]Patil S P, Goswami A, Kalia K, et al. Plant-derived bioactive peptides: A treatment to cure diabetes [J]. Int J Pept Res Ther, 2020, 26: 955.
[31]于國泳. 燕麦、青稞和荞麦抗血小板聚集肽的鉴定及作用的分子机制[D].北京: 北京林业大学, 2015.
[32]王凤. 燕麦多肽的结构特征及DPP4抑制作用[D]. 北京: 北京林业大学, 2016.
[33]董宇婷. 燕麦源α-glucosidase抑制肽与DPP-Ⅳ抑制肽的筛选及抑制机理研究[D]. 哈尔滨: 哈尔滨工业大学, 2019.
[34]Krieger E,Vriend G. YASARA View--molecular graphics for all devices--from smartphones to workstations [J]. Bioinformatics, 2014, 30: 2981.
[35]Van d S D,Lindahl E, Hess B, et al. GROMACS: fast, flexible, and free [J]. J Comput Chem, 2005, 26: 1701.
[36]Weihofen W A, Liu J, Reutter W, et al. Crystal structures of HIV-1 Tat-derived nonapeptides Tat-(1-9)and Trp2-Tat-(1-9)bound to the active site of dipeptidyl-peptidase IV (CD26)[J]. J Biol Chem,2005, 280: 14911.
[37]Maier J A, Martinez C,Kasavajhala K, et al. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB [J]. J Chem Theory Comput, 2015, 11: 3696.
[38]Jorgensen W L, Chandrasekhar J, Madura J D,et al. Comparison of simple potential functions for simulating liquid water[J]. J Chem Phys, 1983, 79: 926.
[39]Hess B,Bekker H, Berendsen H J C, et al. LINCS: a linear constraint solver for molecular simulations[J]. J Chem Theory Comput, 1997, 4: 1463.
[40]Darden T A, York D M, Pedersen L G. Particle mesh Ewald-an N.log(N)method for Ewald sums in large systems[J]. J Chem Phys, 1992, 98: 10089.
[41]Berendsen H J C, Postma J P M, Van Gunsteren W F, et al. Molecular dynamics with coupling to an external bath [J]. J Chem Phys, 1984, 81: 3684.
[42]RMartonák, Laio A, Parrinello M. Predicting crystal structures: the Parrinello-Rahman method revisited [J]. Phys Rev Lett, 2003, 90: 075503.
[43]Valdes-Tresanco M S, Valdes-Tresanco M E, Valiente P A, et al. gmx_MMPBSA: a new tool to perform end-state free energy calculations with GROMACS [J]. J Chem Theory Comput, 2021, 17: 6281.
[44]Verwey E J W. Theory of the stability of lyophobic colloids [J]. J Phys Colloid Chem,1947,51: 631.
[45]Still W C,Tempczyk A, Hawley R C, et al. Semianalytical treatment of solvation for molecular mechanics and dynamics [J]. J Am Chem Soc, 1990, 112: 6127.
引用本文格式:
中 文: 许丽佳,李正文,董宏波,等.燕麦多肽数据库及降血糖活性的虚拟筛选[J]. 四川大学学报: 自然科学版, 2023, 60: 066002.
英 文: Xu L J, Li Z W, Dong H B,et al.Study on the oat peptide database and virtual screening of hypoglycemic activity [J]. J Sichuan Univ: Nat Sci Ed, 2023, 60: 066002.
