基于DNA条形码基因和核基因的美丽小条鳅隐存多样性研究
2023-03-29向登高李跃飞陈蔚涛
高 尚,向登高,李跃飞,李 捷,陈蔚涛
(1.中国水产科学研究院珠江水产研究所,广州 510380;2.上海海洋大学海洋生态与环境学院,上海 201306;3.贵州大学动物科学学院,贵阳 550025)
美丽小条鳅(Micronemacheiluspulcher)隶属于鲤形目(Cypriniformes)条鳅科(Nemacheilidae)小条鳅属(Micronemacheilus),是广泛分布在我国华南地区的小型底栖鱼类[1]。由于受到水利开发、过度捕捞、环境污染等诸多因素的影响,美丽小条鳅的资源量衰退严重,亟需给予关注和保护。运用遗传进化分析手段了解美丽小条鳅的遗传谱系及其空间分布格局对该物种遗传种质资源的保护和管理具有重要实际价值。已有研究表明,小型底栖鱼类扩散能力弱,容易在小范围内形成遗传分化,甚至形成高度分化的遗传谱系或者新种[2,3]。我国华南地区河流网络复杂,拥有珠江水系(包括东江、西江、北江三大干流)、海南岛诸河流及其周边小型陆封型河流,如南流江、漠阳江等。这些河流虽然相隔的地理位置不大,但却相互被海水或者山脉隔离,极大地限制了鱼类群体之间的迁移扩散。另外,华南地区经历的系列地质事件,如海平面波动、山脉隆升等[4,5]也在一定程度上深刻影响着鱼类的进化历史[6]。鉴于上述背景资料,推测美丽小条鳅可能形成了相互独立的遗传谱系甚至新种。丘城锋等[7]和庆宁等[8]分别利用线粒体Cytb基因和控制区片段发现华南西部及海南岛的美丽小条鳅已经形成了多个遗传谱系。然而,由于早期研究涉及研究区域比较有限,并且仅使用了线粒体分子标记,因此很有必要利用高覆盖度的样本和不同类型基因来重新评估美丽小条鳅在遗传层面上的多样性水平以及是否已经进化成多个物种。
DNA条形码作为分类学中辅助物种鉴定的技术,已被广泛应用于物种鉴定[9-11]或隐存种发现的相关研究中[12-14]。针对脊椎动物类群,DNA条形码技术主要通过利用线粒体细胞色素c氧化酶Ⅰ中5′端约650 bp的碱基序列来实现物种的鉴定或者隐存种的挖掘。另外,随着分析方法的改进和完善,越来越多的研究倾向于联合线粒体基因和核基因等多种分子标记进行隐存种的验证[2,15,16]。因此,本研究通过对华南地区多个水系19个站位的美丽小条鳅样本的采集,利用DNA条形码基因(COI基因)和糖基转移酶基因(Glyt基因)评估美丽小条鳅的进化谱系组成,并验证美丽小条鳅是否存在隐存种,旨在为美丽小条鳅后期的管理和保护提供科学支撑。
1 材料与方法
1.1 样品采集
分别于2017年7月-2020年9月进行采集,在珠江水系(东江、西江、北江)、漠阳江、南流江、海南岛的凌水河和南渡江的19个站位(图1)共采集美丽小条鳅样品107尾(表1)。剪取每尾样本的少量胸鳍鳍条并保存在95%的酒精中,带回实验室用以DNA提取。
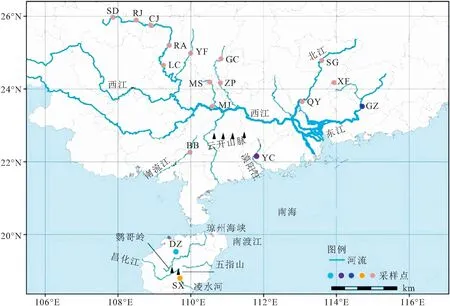
图1 采样示意图
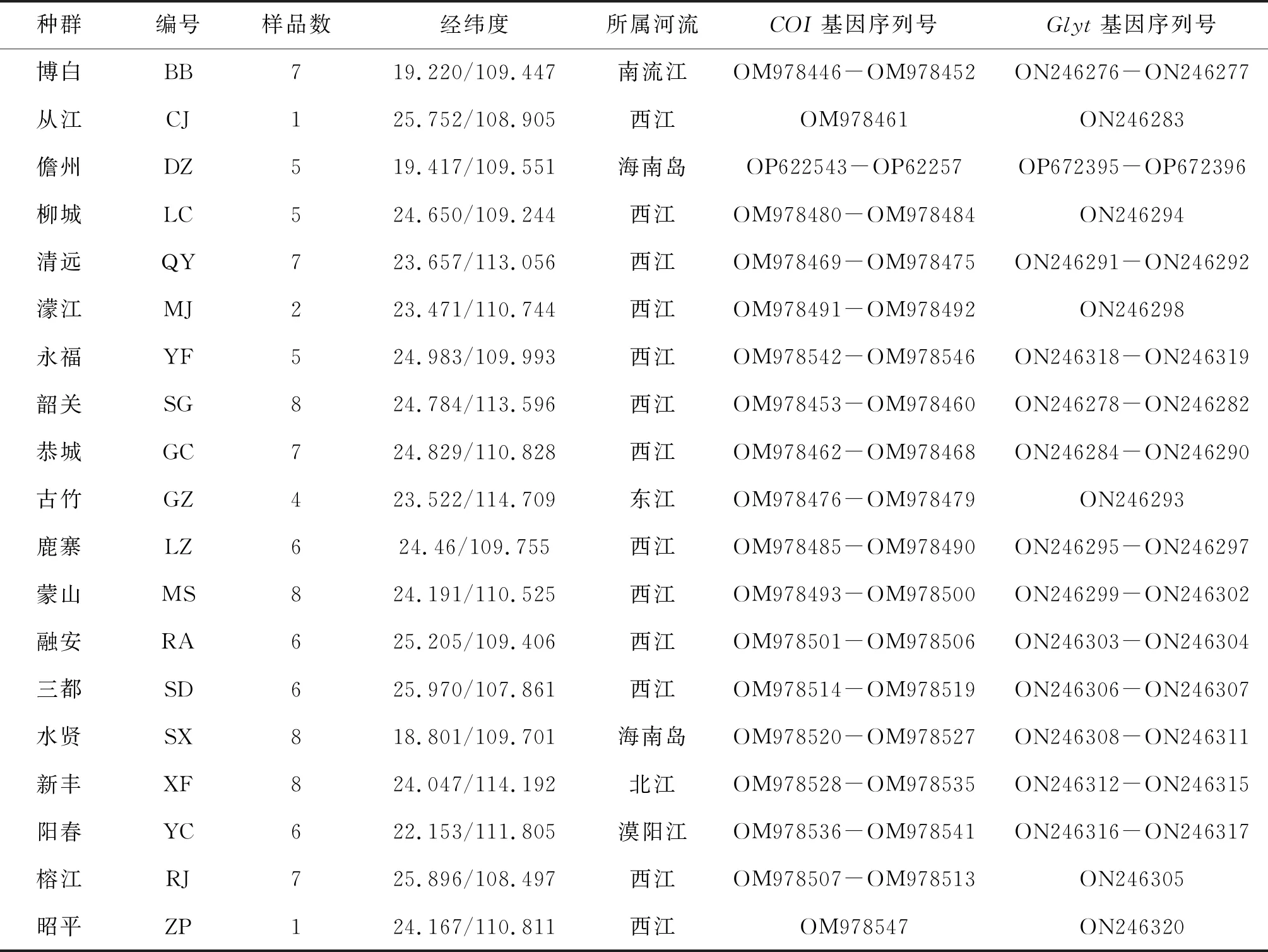
表1 样本采集信息
1.2 基因组DNA提取、扩增与测序
采用高盐法提取基因组总DNA[9]。利用通用引物FishF1(5′-TCAACCAACCACAAAGACATTGGCAC-3′)和FishR1(5′-TAGACTTCTGGGTGGCCAAAGAATCA-3′)[10]扩增线粒体COI基因5′端约650 bp的序列。PCR反应扩增体系为25 μL:2×PCR mix(0.1 U/μL)12.5 μL,正反向引物(10 mmol/L)1 μL,DNA模板1 μL,最后用ddH2O补齐至25 μL。PCR反应条件为:95 ℃预变性5 min,94 ℃变性30 s,54 ℃退火30 s,72 ℃延伸1 min,30个循环,最后再72 ℃延伸10 min。筛选部分样本进行糖基转移酶基因(Glyt)的扩增和测序。使用引物Glyt_F559(5′-GGACTGTCMAAGATGACCACMT-3′)和Glyt_R1562(5′-CCCAAGAGGTTCTTGTTRAAGAT-3′)[11]对Glyt基因进行扩增。PCR反应扩增提取与COI基因相同,反应条件为:95 ℃预变性5 min,95 ℃变性30 s,55 ℃退火1 min,72 ℃延伸1.5 min,35个循环,最后再72 ℃延伸10 min。扩增产物经1%琼脂糖凝胶电泳检测后送往测序公司进行双向测序。
1.3 数据处理与分析
测序获得的DNA序列采用Lasergene v7.1软件包中(DNASTAR,Inc.,USA)的Seqman软件进行人工核查、校正及组装拼接。拼接完成的序列用MEGA 6.06软件[17]进行多重比对,剪切掉冗余片段获得一致序列备用。
利用DnaSP 5.10[18]统计单倍型数,使用MEGA 6.06软件分析序列特征。选取横纹南鳅为外类群,采用邻接法(Neighbor-Joining,NJ)并基于Kimura双参数(K2P)模型[19]构建所有美丽小条鳅COI基因序列邻接关系进化树。另外,使用RaxML-VI-HPC[20]和MrBayes 3.1.2[21]构建基于美丽小条鳅COI基因单倍型数据和Glyt基因等位基因型数据的系统发育树。利用MRMODELTEST 2.3基于赤池信息量原则(AIC)选择最优的核苷酸替代模型(COI:GTR+G;Glyt:K80)。最大似然树自展重复数设置为1 000次;贝叶斯系统发育树运算一共运行了2×108代,每1 000代取样1次,舍弃前25%的运行代数。同时基于不同基因类型,分别利用MEGA估算了不同进化谱系内部和不同进化谱系之间的遗传距离。考虑到核基因进化速率慢的特点,本研究利用POPART 1.7.2绘制了不同谱系之间的等位基因网状图[22],用以查看不同线粒体谱系之间在核基因水平上的进化关系。核基因数据的分型在DnaSP 5.10软件中运行。
利用三种方法对美丽小条鳅物种是否存在隐存物种进行界定。首先,采用Automatic Barcode Gap Discovery(ABGD)[23]基于K2P和JC69距离模型查看了美丽小条鳅的可操作分类单元(Operational taxonomic unit)数目。此外,基于单倍型数据构建的最大似然树,使用Poisson Tree Process(PTP)[24]算法评估美丽小条鳅有效可操作分类单元的数量。最后,利用the General Mixed Yule-coalescent(GMYC)[25]对物种有效性进行评估。因为GMYC分析需要超度量基因树(Ultrametric gene tree),因此利用BEAST version 1.8[26]基于单倍型数据构建了超度量基因树。马尔科夫链长10 000万代,采用GTR+G模型、Speciation:Yule Process 假设,每1 000代建立一棵系统发育树;舍弃前25%后将所有分枝平均后验概率最高者选出,在FigTree中显示最后所得树形图。PTP(http://mptp.h-its.org)和GMYC(http://species.h-its.org/gmyc/)分析分别在网页版服务器上进行运算,参数采取默认设置参数。
为了进一步检验美丽小条鳅不同谱系是否达到物种级别,本研究联合COI基因和Glyt基因,基于贝叶斯系统发育和系统地理的方法(Bayesian Phylogenetics and Phylogeography,BPP)综合评估隐存物种有效性[27]。这个方法考虑了非单系性和不完全谱系分选等因素[27]。分别设置了三组祖先种群规模(ө)和根龄(τ),即ө=(1,10),τ=(1,10);ө=(2,2 000),τ=(2,2 000)和ө=(1,10),τ=(2,2 000)。分析在BPP version 3.0软件中运行,共运行100万代,每5代取样一次,舍弃前5万代。选取基于COI基因单倍型数据获得的系统发育拓扑结构作为引导树。
2 结果
2.1 COI和Glyt序列特征分析
本研究成功获得了107和47尾美丽小条鳅的COI和Glyt基因序列。在序列两端截齐后,共获得610 bp的COI基因序列,其中包含了60个变异位点和55个简约信息位点;909 bp的Glyt基因序列,其中包含了17个变异位点和12个简约信息位点。COI基因和Glyt基因分别界定了24个单倍型和27个等位基因型。
2.2 系统树和遗传距离
基于107尾美丽小条鳅COI基因序列构建的邻接树发现华南地区美丽小条鳅形成了2个进化谱系(Ⅰ和Ⅱ),其中谱系Ⅰ由珠江水系、南流江和漠阳江群体组成(即大陆群体),谱系Ⅱ由海南岛群体组成(图1;图2)。另外,谱系Ⅰ拥有三个子谱系(Ⅰ-1,Ⅰ-2和Ⅰ-3),其中Ⅰ-1主要包括珠江水系的西江和南流江群体,Ⅰ-2和Ⅰ-3分别由珠江水系的东江群体和漠阳江群体组成;谱系Ⅱ含有两个子谱系,分别由陵水河群体和南渡江群体组成。基于COI基因单倍型的最大似然树和贝叶斯树获得了相近的系统发育结构,支持美丽小条鳅存在两个主要谱系(Ⅰ和Ⅱ;图3a)。然后,基于Glyt基因单倍型的最大似然树和贝叶斯树并未获得谱系分化的拓扑结构(图3b),但是均强烈支持大陆群体、海南岛的陵水河群体和南渡江群体均是独立进化的。等位基因型网状图表明基于COI基因获得的谱系Ⅰ和Ⅱ、谱系Ⅱ的两个子谱系Ⅱ-1和Ⅱ-2均不存在共享等位基因型的现象(图4)。

图3 基于美丽小条鳅COI基因单倍型数据(a)和Glyt等位基因型数据(b)的最大似然树和贝叶斯树(仅展示最大似然树)
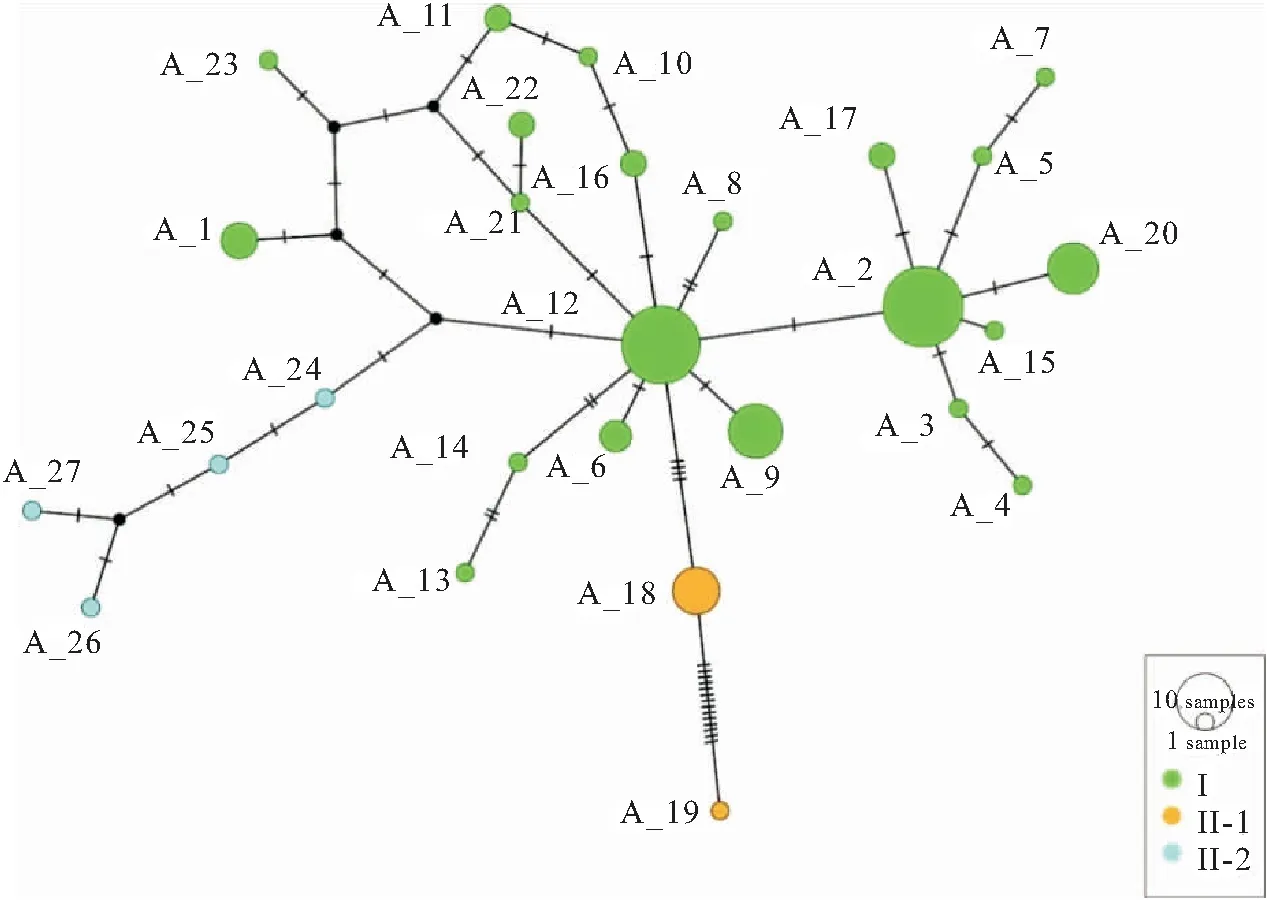
图4 Glyt等位基因型网状图
基于COI基因的美丽小条鳅种内平均遗传距离是1.75%,谱系Ⅰ和Ⅱ内部的平均遗传距离分别是0.61%和1.53%,谱系之间的平均遗传距离是5.83%,其中子谱系Ⅱ-1和Ⅱ-2之间的平均遗传也达到2.92%。基于Glyt基因的美丽小条鳅种内平均遗传距离是0.13%,谱系Ⅰ和Ⅱ内部的平均遗传距离分别是0.06%和0.38%,谱系之间的平均遗传距离是0.37%。
2.3 基于DNA条形码的物种界定
ABGD分析表明当种内差异先验值P在0.001 7~0.035 9之间时均支持3个可操作分类单元,分别是海南岛的陵水河群体和南渡江群体以及珠江-漠阳江-南流江群体(图5)。bPTP分析获得结果与ABGD的结果一致,均支持3个可操作分类单元(图5)。此外,单阈值和多阈值GMYC分析均显示美丽小条鳅存在5个可操作分类单元(图5)。

图5 物种界定分析结果。系统发育树拓扑结构由BEAST构建
2.4 贝叶斯物种界定
贝叶斯系统发育和系统地理(BPP)分析发现基于不同的祖先种群规模(ө)和根龄(τ),DNA条形码基因界定的谱系Ⅰ、谱系Ⅱ的两个子谱系(Ⅱ-1和Ⅱ-2)均获得100%的后验概率,表明这谱系Ⅰ与谱系Ⅱ的两个子谱系可以上升为三个不同物种(图6)。

图6 贝叶斯物种界定结果
3 讨论
3.1 美丽小条鳅多样性的地理分布
已有研究表明,DNA条形码不仅可以实现物种鉴定,还能够划分不同地理种群的分布界限[28-30]。本研究基于不同方法构建的系统发育树均一致显示美丽小条鳅存在两个主要谱系(Ⅰ和Ⅱ),其中谱系Ⅰ和谱系Ⅱ分别包含三个和两个子谱系。丘城锋等[8]和庆宁等[7]关于华南西部和海南岛美丽小条鳅的遗传变异研究也得到了类似的结果。另外,研究发现子谱系Ⅰ-1主要由西江、北江、南流江群体组成,Ⅰ-2由东江群体组成,Ⅰ-3由漠阳江群体组成。谱系Ⅱ包含两个子谱系,其中子谱系Ⅱ-1和Ⅱ-2分别由陵水河群体和南渡江群体组成,呈现严格的地理分布格局(图1和图2)。上述研究结果表明美丽小条鳅拥有丰富的遗传多样性,同时也证实DNA条形码能够用来划分美丽小条鳅不同的地理群体。美丽小条鳅的谱系地理分布格局主要受到华南地区山脉和海峡阻隔导致(图1)。例如,谱系Ⅰ和Ⅱ的分布区被琼州海峡阻隔,子谱系Ⅰ-3与其他两个子谱系的分布区被云开山脉隔离,子谱系Ⅰ-1和Ⅰ-2的分布区被海水阻隔,子谱系Ⅱ-1和Ⅱ-2的分布区被五指山和鹦哥岭阻隔。早期研究也证实华南地区的山脉和海峡塑造了该分布区其他鱼类的遗传多样性地理分布格局,如三角鲂(Megalobramaterminalis)[6]、海南鲌(Culterrecurviceps)[31]、福建纹胸(Glyptothoraxfokiensis)[3]、唐鱼(Tanichthysalbonubes)[15]等。另外,鳅类较弱的扩散能力也是美丽小条鳅形成谱系分化的一个重要因素。研究表明,鳅类营底栖生活,游泳能力弱,容易在局域形成显著的遗传分化甚至进化成多个谱系[32]。考虑到核基因进化速率慢的特点,基于Glyt基因的系统发育树虽然未获得高支持率的拓扑结构,但是等位基因网状图表明大陆群体与海南岛群体、海南岛的南渡江群体(DZ)和陵水河群体(SX)不存在共享等位基因型的现象,表明大陆群体与海南岛群体以及海南岛的陵水河群体与南渡江群体在线粒体和核基因水平上都是独立进化的。
3.2 隐存种
分子测序技术的介入促使越来越多的隐存物种被发现,为生物多样性的保护提供了重要数据[33]。DNA条形码技术的一个重要作用是发现新种或隐存种,并且在近二十年得到了广泛应用[34,35]。本研究发现谱系Ⅰ和谱系Ⅱ之间的平均遗传距离达到了5.64%,子谱系Ⅱ-1和Ⅱ-2之间的平均遗传距离也达到了2.92%,大于DNA条形码研究中物种界定阈值(2%)[36]。基于DNA条形码基因的物种界定分析结果也认为美丽小条鳅至少应该分为三个物种。另外,考虑了非单系性和不完全谱系分选等因素的BPP分析能够综合多个基因信息对不同谱系进行物种有效性的验证,结果也支持美丽小条鳅应该上升为三个独立的物种这个结论。加上严格的地理分布格局,认为华南地区的大陆群体、海南岛的陵水河群体和南渡江群体应该看做三个独立的物种。琼州海峡、五指山与鹦哥岭的地理隔离作用可能是导致美丽小条鳅隐性成种的重要因素。同样的现象在华南地区的福建纹胸[3]和唐鱼[15]也有所体现。例如,CHEN等[3]认为福建纹胸亚种(Glyptothoraxfokiensis)和海南纹胸亚种(Glyptothoraxfokiensishainanensis)应当上升为两个种[8],LI等[15]发现华南地区至少分布有8个唐鱼物种。为寻找更多关于美丽小条鳅隐性成种的证据,后期研究应当开展传统的形态度量学与基于更多分子标记的相关分析。
3.3 保护管理建议
通过分子进化手段揭示物种的遗传多样性的地理分布格局对于物种的精准管理与保护具有重要指导意义。本研究发现美丽小条鳅至少存在三个隐存种,应该当做三个物种有区别开展管理保护工作。另外,谱系Ⅰ拥有三个符合地理分布模式的子谱系,表明它们可能是三个独立进化的种群,可以认为是三个不同的演化显著单位(evolutionary significant unit,ESU)[37]。在未来的保护工作中,这三个演化显著单元应该得到有针对性的管理和保护。由于美丽小条鳅广泛分布于我国华南地区诸多水系江段,考虑到本研究调查站位仅覆盖部分区域,因此该物种的遗传多样性可能被严重低估。为了更为科学地提出保护管理建议,更多样本量、更高覆盖度的相关研究亟待开展。
4 结论
本研究利用DNA条形码COI基因和Glyt基因揭示了华南地区美丽小条鳅的遗传多样性分布以及隐存种问题,发现美丽小条鳅至少存在两个主要进化谱系(Ⅰ和Ⅱ),谱系Ⅰ存在三个具有严格地理分布的子谱系(Ⅰ-1,Ⅰ-2和Ⅰ-3),谱系Ⅱ拥有两个严格地理分布的子谱系(Ⅱ-1和Ⅱ-2)。同时研究证明谱系Ⅰ以及谱系Ⅱ的两个谱系应当看做三个独立物种。本研究认为美丽小条鳅的多样性可能被严重低估,亟需使用形态度量学、高覆盖度的样品和多种类型的分子标记来全面系统揭示其隐藏的遗传多样性,为美丽小条鳅全面精准的管理保护提供数据支持。
