初步探究LncRNA在甘蓝型油菜生态型分化中的作用
2023-03-23杨太桦杨福权郜耿东金庆东徐林珊徐正华葛贤宏周广生
杨太桦 杨福权 郜耿东 殷 帅 金庆东 徐林珊 蒯 婕 汪 波 徐正华 葛贤宏 王 晶 周广生
初步探究LncRNA在甘蓝型油菜生态型分化中的作用
杨太桦 杨福权 郜耿东 殷 帅 金庆东 徐林珊 蒯 婕 汪 波 徐正华 葛贤宏 王 晶*周广生
华中农业大学植物科学技术学院, 湖北武汉 430070
甘蓝型油菜是我国重要的油料作物之一, 根据其成花转变过程中对低温春化时间需求的不同分为3种生态型: 春性、半冬性和冬性。前人研究发现, 长链非编码RNA (LncRNA)可在多层面上调控基因的表达, 参与对植物生长发育的调控。在拟南芥中, LncRNA可以通过调控春化途径相关基因的表达来影响开花。本研究以3种生态型甘蓝型油菜为材料, 利用高通量测序技术进行苗期叶片的mRNA和LncRNA测序, 初步探究LncRNA在油菜生态型分化及适应性形成中的作用。3种生态型甘蓝型油菜共差异表达基因的GO及KEGG富集分析表明, 不同生态型甘蓝型油菜之间存在大量基础化合物合成代谢差异, 特别是脂质类化合物。3种生态型甘蓝型油菜中共鉴定获得3775个LncRNA, 其中285个在2个及以上生态型组合中存在差异表达, 涉及到1517个候选靶基因。这些差异表达LncRNA涉及到的靶基因也富集到大量基础化合物合成代谢途径。通过mRNA-LncRNA联合分析, 我们预测到了一个开花基因的调控网络, 包含8个开花基因和23个LncRNA, 涉及到温度和光信号调控通路。通过比较鉴定得到的LncRNA和972个甘蓝型油菜重要农艺性状QTL位置信息发现约90%的LncRNA位点和QTL区间存在重叠, 且差异表达LncRNA和QTL的重叠位点在不同生态型油菜中的分布具有差异。结果说明LncRNA在油菜生态型分化及重要农艺性状形成中具有重要作用。
甘蓝型油菜; 生态型; 长链非编码RNA; 开花基因; 农艺性状位点
甘蓝型油菜(L., 2= 38)是我国及世界上重要的油料作物之一, 菜籽油约占我国国产食用植物油的1/3[1]。由于自然选择和人工选择驯化, 甘蓝型油菜形成了适应不同气候和纬度区域种植的生态类型。根据其成花转化中对低温春化需求的不同分为冬油菜(winter oilseed rape, WOSR)、半冬性油菜(semi-winter oilseed rape, SWOSR)和春油菜(spring oilseed rape, SOSR)。冬油菜是最原始的类型, 于欧洲中世纪晚期开始种植, 开花晚, 需要严格的春化[2]; 春油菜在1700年左右形成, 其开花早, 无需春化[3]; 此外, 甘蓝型油菜于1930年前后传入中国, 经过选择后产生了新的生态类型, 即为半冬性油菜, 需要适当的春化[3-4]。
作物的开花时间是重要的农艺性状之一, 受外界环境因子和自身发育信号共同调控。在拟南芥中有超过300个开花相关的基因被鉴定[5]。随着测序技术的发展及多个高质量甘蓝型油菜参考基因组的公布(Darmor-bzh、宁油7号、中双11号), 研究人员发现基因是影响甘蓝型油菜春化需求和开花时间的重要位点[6-9]。基于自然群体的重测序及关联分析研究中, 研究人员鉴定到大量的与生态型分化/开花时间显著关联的位点, 也发现基因对于生态型分化起到了重要作用[2,10-13]。An等[14]通过群体转录组学数据也挖掘到了一些开花或调节春化的基因。Song等[15]完成了甘蓝型油菜泛基因组构建, 并且通过巢式关联群体进行了全基因组关联分析, 鉴定到67个开花候选基因, 进一步证实基因等位变异在一定程度上决定了油菜对春化的需求及开花时间。Yin等[11]则通过等位基因序列分离及转基因验证, 进一步发现与共同调控不同类型甘蓝型油菜对春化的需求。与上述研究相一致, 有研究认为甘蓝型油菜9个基因可能均参与了油菜春化需求的调节[16-18]。
非编码RNA是一种不具有编码蛋白质能力的RNA, 长期以来被认为是无用的转录“噪音”[19]。长链非编码RNA (LncRNA)作为非编码RNA的一种, 是长度大于200个核苷酸的RNA, 普遍存在于动植物基因组中, 参与细胞分化及个体发育等一系列生物学过程。前人研究发现, 在拟南芥中LncRNA作为重要的转录调控因子调控基因, 从而调控开花进程[20]。目前为止, 甘蓝型油菜中有关LncRNA的研究还不多见。Song等[21-22]最早鉴定到了一条花粉特异性的非编码RNA, 其参与甘蓝型油菜花粉发育的调控。Joshi等[23]在甘蓝型油菜中鉴定到3181个LncRNA, 发现一个LncRNA可能参与了核盘菌防御响应。Zhang等[24]分别在甘蓝型油菜中鉴定到1885个LncRNA, 发现LncRNA表现出了A亚基因组倾向性的表达。Shen等[25]发现LncRNA可能在脂质代谢中发挥作用, 可能参与了对编码oleosin1蛋白基因的调控, 进而参与种子脂质的积累过程。Tan等[26]的研究发现, 干旱胁迫和复水处理影响了部分LncRNA的表达, 表明LncRNA存在响应应激的功能。
本研究基于第二代高通量测序技术, 对3种生态型甘蓝型油菜进行了LncRNA-seq和mRNA-seq分析, 并结合已报道的重要农艺性状的QTL信息, 初步探究LncRNA在甘蓝型油菜生态分化及适应性形成中的作用, 为油菜遗传育种以及适应性改良提供理论依据。
1 材料与方法
1.1 试验材料与测序数据
本研究中用到的甘蓝型油菜品种为Westar (春油菜)、Dunkeld (春油菜)、Tapidor (冬油菜)、Darmor (冬油菜)、中双11号(半冬性油菜)和宁油7号(半冬性油菜), 材料于2017—2018年种植于华中农业大学试验田。待6种甘蓝型油菜都生长至五片真叶期时, 同时取第3片幼嫩叶片用于总RNA的提取用于建库测序, 每个材料各2个生物学重复,-80℃保存备用。
使用高纯总RNA快速提取试剂盒(北京百泰克生物技术有限公司)进行RNA的提取, cDNA反转引物为随机六引物, 建库试剂盒为TruSeq Stranded Total RNA with Ribo-Zero Plant (Illumina)。LncRNA、mRNA建库和测序均由华中农业大学作物遗传改良国家重点实验室高通量测序平台完成。为更好地鉴定LncRNA, 本研究选用组装质量较好的v10 (contig N50 = 11.49 Mb)[27]为参考基因组进行后续的分析。
1.2 转录组数据分析
原始下机数据通过FastQC (version = 0.11.5)[28]软件进行质量检测, 并使用Trimmomatic (version = 0.39)[29]软件去接头序列和除低质量reads。使用Hisat2 (version = 2.1.0)[30]软件将质控后的clean reads比对到参考基因组上, 使用StringTie (version = 2.1.4)[31]获取每个基因上的reads数量并用TPM (transcripts per kilobase of exon model per million mapped reads, 每千个碱基的转录每百万映射读取的转录本数)表示基因的表达量。使用Deseq2 (version = 3.3.3)[32]软件进行基因差异表达分析, 筛选标准为 |log2(Fold Change) | ≥ 2且adj< 0.01。
1.3 LncRNA数据分析
原始下机数据通过FastQC软件进行质量检测, 并使用Trimmomati软件去除低质量的reads, 然后使用Hisat2软件将质控后的clean reads比对到参考基因组上。使用StringTie软件进行有参组装, 得到组装的gtf文件。使用gffcompare (version = v0.12.2)[33]将得到的gtf文件与参考基因组gff注释文件进行比较, 提取标记为i、j、o、u和x的转录本序列。对得到的转录本进行过滤: (1) 保留转录本长度≥200 bp、表达量TPM≥1以及外显子数目 > 1的转录本。(2)使用CPC2[34]、CPAT[35]和PLEK[36]软件预测转录本编码能力, 取交集保留不具有编码能力的转录本。将最终得到的LncRNA转录本比对到LncRNA数据库-CANTATAdb (http://yeti.amu.edu.pl/CANTATA)[37], 没比对上的视为新鉴定的LncRNA。使用Deseq2软件进行基因差异表达分析, 筛选标准为 |log2(Fold Change)|≥2且adj< 0.01。
2 结果与分析
2.1 3种生态型油菜的开花时间具有差异
在半冬性环境条件下, 春油菜开花所需时间最短, 其次是半冬性油菜, 冬油菜开花所需的时间最长(2014年武汉: Westar: 146 d、Dunkeld: 157 d、中双11号: 158 d、宁油7号: 152 d、Tapidor: 179 d、Darmor: 181 d); 在春环境条件下, 冬油菜基本无法开花, 春油菜开花所需时间短于半冬性油菜, 并且他们的开花所需时间为在半冬性环境下的一半(2013年青海: Westar: 56 d、Dunkeld: 61 d、中双11号: 78 d、宁油7号: 70 d、Tapidor和Darmor不开花; 2014年甘肃: Westar: 51 d、Dunkeld: 67 d、中双11号: 70 d、宁油7号: 65 d、Tapidor和Darmor不开花)[4,10,15]。在武汉半冬性环境条件下, 2021—2022年度, Westar (春油菜)、中双11号(半冬性油菜)和Tapidor (冬油菜)的开花时间分别为107、162和189 d。播种后110 d, 3种油菜外观形态也存在较大差别(图1)。

图1 3种生态型甘蓝型油菜同时期形态
在武汉半冬性环境下, 2021-2022年度, 播种约110 d时, 3种生态型甘蓝型油菜形态差异明显。A: Westar; B: 中双11号; C: Tapidor。标尺为20 cm。
Morphology of three ecotypes of, about 110 days after sowing in semi-winter environment of Wuhan, in 2021-2022. A: Westar; B: Zhongshuang 11; C: Tapidor. Bar: 20 cm。
2.2 甘蓝型油菜全基因表达谱
为探究不同生态型甘蓝型油菜之间的基因表达情况, 本研究对3种生态型共6种甘蓝型油菜品种进行转录组测序, 按照3种生态型进行分组分析。将表达量TPM≥1的基因视为存在表达的基因, 经过过滤, 每个样本mRNA-seq产生了168.42~256.30百万条clean reads。12个甘蓝型油菜mRNA-seq数据比对率为92.96%~98.00%。主成分分析表明同组样本之间具有很好的一致性(图2-A)。总体而言, 甘蓝型油菜中, 40.45%~44.75%的基因存在表达, 37.26% (40,308)的基因在春油菜中表达, 39.22% (42,429)的基因在冬油菜中表达, 38.56% (41,713)的基因在半冬性油菜中表达(图2-B)。32.52% (35,184)的基因在3种生态型甘蓝型油菜中均表达, 51.82% (56,066)的基因至少在一种甘蓝型油菜中表达(图2-B)。2843、2629和2225个差异表达基因分别在春油菜与冬油菜、春油菜与半冬性油菜和半冬性油菜与冬油菜之间检测到(图2)。其中27个基因在3种生态型组合中均鉴定为差异表达基因(图2-D)。

图2 3种生态型6个甘蓝型油菜品种mRNA-seq分析相关结果
A: 6种甘蓝型油菜品种12个样本mRNA-seq主成分分析; B: 在6个甘蓝型油菜品种和3种生态型中甘蓝型油菜全部基因表达情况; C: 甘蓝型油菜3种生态型两两之间差异表达基因数量; D: 甘蓝型油菜3种生态型两两之间差异表达基因韦恩图。SOR: 春油菜; SWOR: 半冬性油菜; WOR: 冬油菜。
A: mRNA-seq PCA analysis of 12 samples from sixcultivars; B: the relative expression level of all genes in sixcultivars and three ecotypes; C: the number of differentially expressed genes among three ecotypes; D: Venn diagram of differentially expressed genes among three ecotypes of. SOR: spring type rapeseed; SWOR: semi-winter type rapeseed; WOR: winter-type rapeseed.
2.3 不同生态型甘蓝型油菜差异表达基因分析
为探究这些差异基因在不同生态型中的参与的功能, 本研究对在2组及以上生态型中出现差异表达的基因(mRNA-DEG, 共2300个基因)进行GO和KEGG富集分析。GO富集分析表明, 这些基因显著富集在脂质定位(GO:0010876)、脂质转运(GO:0006869)、磷脂酶C活性(GO:0004629)、磷脂酰肌醇磷脂酶C活性(GO:0004435)、磷酸二酯水解酶活性(GO:0008081)、磷脂酶活性(GO:0004620)等通路(图3-A)。KEGG富集分析表明, 这些基因显著富集在黄酮类物质生物合成、苯丙烷类物质生物合成、异黄酮类生物合成、α-亚麻酸代谢等通路(图3-B)。表明不同生态型甘蓝型油菜的分化形成可能涉及到了大量基础化合物的合成代谢, 并且脂质类相关化合物可能发挥了重要的作用。

图3 3种生态型甘蓝型油菜差异表达基因GO功能和KEGG通路富集分析
A: 3种生态型甘蓝型油菜差异表达基因GO功能富集分析; B: 3种生态型甘蓝型油菜差异表达基因KEGG通路富集分析。
A: GO function enrichment of differentially expressed genes in three ecotypes of; B: KEGG pathway enrichment of differentially expressed genes in three ecotypes of.
2.4 LncRNA鉴定和靶基因鉴定分析
在之前的研究中, 报道了lncRNA作为重要的转录调控因子调控植物的开花[20]。本研究对3种生态型6种甘蓝型油菜品种进行LncRNA测序分析。每个样本LncRNA-seq产生了157.31~537.70百万条clean reads。12组甘蓝型油菜LncRNA-seq数据比对率为93.30%~95.67%。用Stringtie软件对LncRNA测序数据进行组装, 12组样本中共得到了146,188条不同的转录本, 过滤后共鉴定得到3775个LncRNA序列, 其中97.91% (3696)的LncRNA可以定位在甘蓝型油菜19条染色体上。主成分分析表明同组样本之间的LncRNA表达量具有很好的一致性(图4-A)。将鉴定到的甘蓝型油菜LncRNA序列与白菜、甘蓝和甘蓝型油菜LncRNA数据库进行比对分析, 分别比对上了1190、1418和2176个LncRNA, 另外鉴定到1147个新LncRNA序列。28.90% (1091), 26.86% (1014)和28.32% (1069)的LncRNA分别在春油菜、半冬性油菜和冬油菜中表达(图4-B)。其中, 688个LncRNA在春油菜与冬油菜中都表达; 645个在半冬性油菜与春油菜中表达; 640个在冬油菜与半冬性油菜中表达。522 (13.83%)个LncRNA在3种生态型甘蓝型油菜中都表达(图4-C)。在油菜19条染色体上, A10染色体上鉴定到的LncRNA数量最少(79), C03上数量最多(307)。C亚基因组上LncRNA数量要远多于A亚基因组(A亚基因组: 1486, C亚基因组: 2210), 这可能与甘蓝型油菜参考基因组A、C 亚基因组实际大小和基因数量有关(A亚基因组大小: 346M, 基因数量: 47,193; C亚基因组大小: 520M, 基因数量: 59,692)(图4-D)。
图4 3种生态型6个甘蓝型油菜品种LncRNA-seq分析相关结果
A: 6种甘蓝型油菜品种12个样本LncRNA-seq主成分分析; B: 在6个甘蓝型油菜品种和3种生态型中存在表达的LncRNA数量; C: 甘蓝型油菜3种生态型之间LncRNA表达情况韦恩图; D: 在3种生态型甘蓝型油菜中共鉴定到了LncRNA在19条染色体以及An/Cn亚基因组上的分布。SOR: 春油菜; SWOR: 半冬性油菜; WOR: 冬油菜。
A: LncRNA-seq PCA analysis of 12 samples from sixcultivars; B: the number of expressed LncRNA in sixcultivars and three ecotypes; C: Venn diagram of expressed LncRNA among three ecotypes; D: the distribution of LncRNA on 19 chromosomes and An, Cn subgenomes in three ecotypes of. SOR: spring type rapeseed; SWOR: semi-winter type rapeseed; WOR: winter-type rapeseed.
2.5 不同生态型甘蓝型油菜LncRNA差异表达分析
按照生态型, 本研究对LncRNA进行了差异表达分析。在春油菜和冬油菜、春油菜和半冬性油菜、半冬性油菜和冬油菜中分别检测到291、352和285个差异表达LncRNA序列。其中有283种LncRNA序列在多个组合中存在表达差异。LncRNA可以通过调控mRNA的表达来实现功能, 本研究通过其与蛋白质编码基因的位置关系(顺式)和表达相关性(反式)预测LncRNA可能参与的生物学功能。预测得到1517个预测靶基因(顺式: 617, 反式: 911, 11个预测靶基因在顺式和反式预测中均检测到), 并对这些表达差异LncRNA的预测靶基因(LncRNA-DEG)进行GO和KEGG分析。对于GO功能富集分析表明, 差异表达LncRNA预测候选靶基因富集在众多跨膜转运蛋白活性(如: 含核碱基的化合物, GO:0015932、碳水化合物衍生物, GO:1901505、有机磷酸酯: GO:0015605等),同样也显著富集在脂质定位(GO:0010876)、脂质转运(GO:0006869)通路(图5-A)。KEGG通路分析富集到DNA复制, 花生四烯酸代谢,错配修复, 氮素代谢以及一些氨基酸萜类物质的合成代谢(图5-B)。

图5 3种生态型甘蓝型油菜差异表达LncRNA候选靶基因GO功能和KEGG通路富集分析
A: 3种生态型甘蓝型油菜差异表达LncRNA候选靶基因GO功能富集分析; B: 3种生态型甘蓝型油菜差异表达LncRNA候选靶基因KEGG通路富集分析。
A: GO function enrichment of candidate target gene of differentially expressed LncRNA in three ecotypes of; B: KEGG pathway enrichment of candidate target gene of differentially expressed LncRNA in three ecotypes of.
2.6 甘蓝型油菜开花基因与LncRNA、mRNA之间的关系
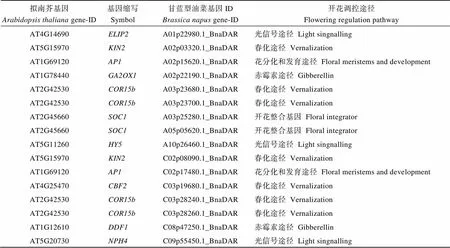
根据拟南芥中鉴定获得的300多个开花时间相关基因[5], 使用双向BLASTP方法在甘蓝型油菜_v10基因组中得到同源基因1165个。将上述mRNA-seq与LncRNA-seq数据进行整合分析, 结果发现开花基因中有39个基因为差异表达基因, 包括、、等基因(附表1)。其中, 有16个基因为LncRNA差异表达预测的候选靶基因, 包括、、和(附表2)。特别是, 11个开花相关基因同时为差异表达基因和LncRNA差异表达预测候选靶基因,、、、、和(附表3)。这11个共差异基因聚类到4个mRNA-LncRNA互作模块, 其中最大的一个大模块包含8个基因和23个LncRNA序列(图6)。其中包含基因的4个拷贝(A03p23680.1_BnaDAR、A03p23700.1_BnaDAR、C03p28240.1_BnaDAR、A03p28260.1_BnaDAR)、基因的2个拷贝(A02p03320.1_BnaDAR、C02p08090.1_BnaDAR)、1个基因(A05p05620.1_ BnaDar)和1个基因(A01p22980.1_BnaDAR)。此外, 本研究用已经发表的甘蓝型油菜群体转录组学数据[38]进行了验证, 该模块中的绝大多数基因在3种生态型油菜群体中存在表达差异。
2.7 LncRNA与甘蓝型油菜农艺性状QTL的位置关系
本研究收集了972个甘蓝型油菜农艺性状QTL[39](附表4), 利用BEDTools软件将鉴定到的LncRNA与972 (A亚基因组: 624; C亚基因组: 348)个QTL的物理区间进行比较。发现在972个QTL位点置信区间内, 其中的844个QTL区间中共有1068个LncRNA位点存在(28.29%, A亚基因: 572; C亚基因: 496) (图7)。结合前面的差异表达LncRNA分析, 我们发现分别有31.96% (93)、30.97% (109)和30.18% (86)与农业性状QTL位点重合的LncRNA分别为春油菜-冬油菜、春油菜-半冬性油菜和半冬性油菜-冬性油菜差异表达LncRNA。与性状QTL位点重合的LncRNA, 更多的分布在甘蓝型油菜的A亚基因组上, 这可能是由于更多的农艺性状相关的QTL在A亚基因组上被检测到。但有趣的是, 在与性状QTL位点重合的LncRNA中, A亚基因组中检测到了更高比例的差异表达LncRNA (春油菜-冬油菜、春油菜-半冬性油菜和半冬性油菜-冬油菜A亚基因组中分别为8.92%、12.76%和8.39%, 对应的C亚基因组为: 8.47%、7.26%和7.66%)。这意味着A亚基因组在甘蓝型油菜生态型分化上起到了很大部分的作用。
3 讨论
目前为止, 3种生态型甘蓝型油菜都有至少2个品种完成了高质量参考基因组的构建, 这些基因组数据的释放为重要农艺性状的基因定位及其功能研究提供了极大的便利[6-9,15]。甘蓝油菜不同生态类型, 不仅开花时间的早晚差别显著, 在形态及重要农艺性状上也有显著不同。尽管不同调控途径的开花基因在拟南芥中都已经研究的很透彻, 但对于经历了多次的基因组多倍化事件的甘蓝型油菜而言, 由于基因拷贝数的增加及功能分化, 鉴定决定其生态型分化、开花时间及其他农艺性状的关键基因仍然具有挑战性。通过双向BLASTP方法和严格的筛选过滤, 本研究在甘蓝型油菜Darmor-bzh v10参考基因组中得到1165个甘蓝型油菜开花基因, 这与之前的报导的1173个基因数量较为接近[40]。
基于mRNA-seq分析, 本研究在不同生态型甘蓝型油菜中检测到了5个同源基因(A02p00340.1_ BnaDAR、A03p16730.1_BnaDAR、C03p04920.1_ BnaDAR、C09p67350.1_BnaDAR、C09p67380.1_ BnaDAR)和2个(A05p05620.1_BnaDAR、C03p30160.1_BnaDAR)出现了差异表达(附表1), 但是没有鉴定到成花素基因, 这可能是因为基因在开花之前表达量极低。在模式植物拟南芥中, 多个开花途径, 包括环境响应和发育信号, 都汇集于对基因的转录调控。基因的表达由光周期途径中基因激活, 并且受到基因的抑制,表达的产物同时受到春化途径和自主途径的抑制[41-42]。

图6 差异表达mRNA-LncRNA互作模块及基因在甘蓝型群体中的达量
A: 差异表达mRNA-LncRNA互作模块, 包含8个基因和23个LncRNA序列; B: 差异表达mRNA-LncRNA互作模块中,、和基因(多个拷贝)在甘蓝型油菜不同生态型群体中的表达量。FPKM (Fragments Per Kilobase of exon model per Million mapped fragments): 每千个碱基的转录每百万映射读取的片段数, *表示在< 0.05水平差异显著, **表示在< 0.01水平差异显著, ns表示差异不显著> 0.05。SOR: 春油菜, SWOR: 半冬性油菜, WOR: 冬油菜。
A: the differentially expressed mRNA-LncRNA interaction module, including 8 genes and 23 LncRNA sequences; B: the relative expression level of COR15b, KIN2, and SOC1 genes (multiple copies) in different ecotypes ofpopulation. FPKM: the fragments per kilobase of exon model per million mapped fragments; * indicates significant differences at the 0.05 probability level; ** indicates significant differences at the 0.01 probability level; ns: no significant difference (> 0.05). SOR: spring type rapeseed; SWOR: semi-winter type rapeseed; WOR: winter-type rapeseed.

图7 甘蓝型油菜QTL和鉴定到的LncRNA共线性分布
植物中第一个被鉴定的LncRNA是黄瓜中的CR20[43], 它是一个细胞分裂素抑制基因。在拟南芥中具有代表性的且通过功能验证的是名为COOLAIR的反义转录本(NAT), 其在低温处理14 d的条件下高表达, 通过抑制基因表达从而促进开花[17,41]。在白菜中经过低温处理后在基因也鉴定到反义转录本的存在, 并且同样能抑制基因的表达[44]。本研究中, 并没有鉴定到以为靶基因的LncRNA序列, 但是检测到了19个LncRNA序列涉及到3个靶基因。本研究鉴定得到了一个包含8个开花相关基因和23个LncRNA的互作表达模块, 其中包含4个基因、2个基因、1个基因和1个基因(图6)。其中编码一种在冷胁迫过程中保护叶绿体膜的蛋白质[45],编码一个可被低温和脱落酸诱导的蛋白[46], 可能与低温驯化有关。编码一种光诱导蛋白, 响应光信号[47],则编码开花途径中的整合因子, 调控及其他基因表达[48]。因此, 该模块可能参与通过响应光信号和温度信号调控甘蓝型油菜的开花时间, 但具体的功能还待进一步探究。目前为止有关LncRNA的功能研究还较少。有研究表明, LncRNA可能通过调节开花相关基因在花发育过程中的表达从而影响鹰嘴豆的产量[50]; LncRNA可能介导植物体对营养元素胁迫响应[51-52]; LncRNA可能参与对病原体以及干旱等逆境胁迫的调控[53-54]。本研究的相关结果为解析LncRNA油菜生态型分化、适应性及、农艺性状的形成中的作用奠定了基础。
多项研究表明, 甘蓝型油菜A亚基因组变异程度、核苷酸多样性、基因组互作程度和连锁不平衡衰退都要强于C亚基因组[2-3,14,49]。大量的GWAS分析也表明, 甘蓝型油菜重要农艺性状QTL多在A亚基因组中, A亚基因组在甘蓝型油菜形成、进化和驯化中起到了关键作用。本研究也发现与性状QTL位点重合的LncRNA, 更多的分布在甘蓝型油菜的A亚基因组上, 且A亚基因组中也检测到了更高比例的差异表达LncRNA。这些结果预示LncRNA可能参与了甘蓝型油菜的生态型分化及与之相适应的农艺性状形成的调控, 且与C基因组相比, A亚基因组在其中扮演了更为重要的角色。
必须指出, 为兼顾结果的准确可靠及研究的经济性, 本研究中每种甘蓝型油菜的转录组测序只进行了2次生物学重复。尽管差异表达的基因及差异表达的LncRNA均是按照不同生态型分组进行的比较, 即每种生态型包含2个品种、4个生物学重复, 但由于同一个生态型的2个品种遗传背景差异较大, 可能会缩小不同生态型间的共差异基因及生态型保守LncRNA数量。
4 结论
本研究通过高通量测序技术, 在3种生态型6个甘蓝型油菜品种中进行mRNA-seq和LncRNA-seq,探究LncRNA在甘蓝型油菜生态型分化中的潜在作用。本研究鉴定到了3775条LncRNA序列, 其中新鉴定到1147条LncRNA序列, 预测到了8575个LncRNA候选靶基因。基于差异表达基因及LncRNA靶基因的GO和KEGG富集分析表明, LncRNA可能参与到氨基酸、脂质和酶等相关基础物质的合成代谢。特别是, 还鉴定到一个包含开花相关基因和LncRNA的互作模块, 可能参与光信号及和温度响应过程。结合与QTL的共定位分析, 进一步发现LncRNA可能在甘蓝型油菜生态型分化、开花期调控及农艺性状形成中发挥重要作用。
[1] 王汉中, 殷艳. 我国油料产业形势分析与发展对策建议. 中国油料作物学报, 2014, 36: 414–421.
Wang H Z, Yin Y. Analysis and strategy for oil crop industry in China., 2014, 36: 414–421 (in Chinese with English abstract).
[2] Lu K, Wei L J, Li X L, Wang Y T, Wu J, Liu M, Zhang C, Chen Z Y, Xiao Z C, Jian H J, Cheng F, Zhang K, Du H, Cheng X C, Qu C M, Qian W, Liu L Z, Wang R, Zou Q Y, Ying J M, Xu X F, Mei J Q, Liang Y, Chai Y R, Tang Z L, Wan H F, Ni Y, He Y J, Lin N, Fan Y H, Sun W, Li N N, Zhou G, Zheng H K, Wang X W, Paterson A H, Li J N. Whole-genome resequencing revealsorigin and genetic loci involved in its improvement., 2019, 10: 1154.
[3] Hu D D, Jing J J, Snowdon R J, Mason A S, Shen J, Meng J L, Zou J. Exploring the gene pool ofby genomics-based approaches., 2021, 19: 1693–1712.
[4] Wei D Y, Cui Y X, He Y J, Xiong Q, Qian L W, Tong C B, Lu G Y, Ding Y J, Li J N, Jung C, Qian W. A genome-wide survey with different rapeseed ecotypes uncovers footprints of domestication and breeding., 2017, 68: 4791–4801.
[5] Bouché F, Lobet G, Tocquin P, Périlleux C. FLOR-ID: an interactive database of flowering-time gene networks in., 2016, 44: D1167–D1171.
[6] Chalhoub B, Denoeud F, Liu S Y, Parkin I A P, Tang H B, Wang X Y, Chiquet J, Belcram H, Tong C B, Samans B, Corréa M, Silva C D, Just J, Falentin C, Koh C S, Clainche I L, Bernard M, Bento P, Noel B, Labadie K, Alberti A, Charles M, Arnaud D, Guo H, Daviaud C, Alamery S, Jabbari K, Zhao M X, Edger P P, Chelaifa H, Tack D, Lassalle G, Mestiri I, Schnel N, Paslier MC L, Fan G Y, Renault V, Bayer P E, Golicz A A, Manoli S, Lee T H, Thi V H D, Chalabi S, Hu Q, Fan C C, Tollenaere R, Lu Y H, Battail C, Shen J X, Sidebottom C H D, Wang X F, Canaguier A, Chauveau A, Bérard A, Deniot G, Guan M, Liu Z S, Sun F M, Lim Y P, Lyons E, Town C D, Bancroft I, Wang X W, Meng J L, Ma J X, Pires J C, King G J, Brunel D, Delourme R, Renard M, Aury J M, Adams K L, Batley J, Snowdon R J, Tost J, Edwards D, Zhou Y M, Hua W, Sharpe A G, Paterson A H, Guan C Y, Wincker P. Early allopolyploid evolution in the post-neolithicoilseed genome., 2014, 345: 950–953.
[7] Sun F M, Fan G Y, Hu Q, Zhou Y M, Guan M, Tong C B, Li J N, Du D Z, Qi C K, Jiang L C, Liu W Q, Huang S M, Chen W B, Yu J Y, Mei D S, Meng J L, Zeng P, Shi J Q, Liu K D, Wang X, Wang X F, Long Y, Liang X M, Hu Z Y, Huang G D, Dong C H, Zhang H, Li J, Zhang Y L, Li L W, Shi C C, Wang J H, Lee S M Y, Guan C Y, Xu X, Liu S Y, Liu X, Chalhoub B, Hua W, Wang H Z. The high-quality genome ofcultivar ‘ZS11’ reveals the introgression history in semi-winter morphotype., 2017, 92: 452–468.
[8] Zou J, Mao L F, Qiu J, Wang M, Jia L, Wu D Y, He Z S, Chen M H, Shen Y F, Shen E H, Huang Y J, Li R Y, Hu D D, Shi L, Wang K, Zhu Q H, Ye C Y, Bancroft I, King G J, Meng J L, Fan L J. Genome-wide selection footprints and deleterious variations in young Asian allotetraploid rapeseed., 2019, 17: 1998–2010.
[9] Chen X Q, Tong C B, Zhang X T, Song A X, Hu M, Dong W, Chen F, Wang Y P, Tu J X, Liu S Y, Tang H B, Zhang L S. A high-qualitygenome reveals expansion of transposable elements, subgenome evolution and disease resistance., 2021, 19: 615–630.
[10] Wu D Z, Liang Z, Yan T, Xu Y, Xuan L J, Tang J, Zhou G, Lohwasser U, Hua S J, Wang H Y, Chen X Y, Wang Q, Zhu L, Maodzeka A, Hussain N, Li Z L, Li X M, Shamsi I H, Jilani G, Wu L D, Zheng H K, Zhang G P, Chalhoub B, Shen L S, Yu H, Jiang L X. Whole-genome resequencing of a worldwide collection of rapeseed accessions reveals the genetic basis of ecotype divergence., 2019, 12: 30–43.
[11] Yin S, Wan M, Guo C C, Wang B, Li H T, Li G, Tian Y Y, Ge X H, King G J, Liu K D, Li Z Y, Wang J. Transposon insertions within alleles ofandare associated with seasonal crop type in rapeseed., 2020, 71: 4729–4741.
[12] Kang L, Qian L W, Zheng M, Chen L Y, Chen H, Yang L, You L, Yang B, Yan M L, Gu Y G, Wang T Y, Schiessl S V, An H, Blischak P, Liu X J, Lu H F, Zhang D W, Rao Y, Jia D H, Zhou D G, Xiao H G, Wang Y G, Xiong X H, Mason A S, Pires J C, Snowdon R J, Hua W, Liu Z S. Genomic insights into the origin, domestication and diversification of., 2021, 53: 1392–1402.
[13] Hu J H, Chen B Y, Zhao J, Zhang F G, Xie T, Xu K, Gao G Z, Yan G X, Li H G, Li L X, Ji G X, An H, Li H, Huang Q, Zhang M L, Wu J F, Song W L, Zhang X J, Luo Y J, Pires J C, Batley J, Tian S L, Wu X M. Genomic selection and genetic architecture of agronomic traits during modern rapeseed breeding., 2022, 54: 694–704.
[14] An H, Qi X S, Gaynor M L, Hao Y, Gebken S C, Mabry M E, McAlvay A C, Teakle G R, Conant G C, Barker M S, Fu T D, Yi B, Pires J C. Transcriptome and organellar sequencing highlights the complex origin and diversification of allotetraploid., 2019, 10: 2878.
[15] Song J M, Guan Z L, Hu J L, Guo C C, Yang Z Q, Wang S, Liu D X, Wang B, Lu S P, Zhou R, Xie W Z, Cheng Y F, Zhang Y T, Liu K D, Yang Q Y, Chen L L, Guo L. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of., 2020, 6: 34–45.
[16] Calderwood A, Lloyd A, Hepworth J, Tudor E H, Jones D M, Woodhouse S, Bilham L, Chinoy C, Williams K, Corke F, Doonan J H, Ostergaard L, Irwin J A, Wells R, Morris R J. Totaltranscript dynamics from divergent paralogue expression explains flowering diversity in., 2021, 229: 3534–3548.
[17] Akter A, Itabashi E, Kakizaki T, Okazaki K, Dennis E S, Fujimoto R. Genome triplication leads to transcriptional divergence ofgenes during vernalization in the genus., 2021, 11: 619417.
[18] Schiessl S V, Quezada-Martinez D, Tebartz E, Snowdon R J, Qian L W. The vernalisation regulatoris differentially expressed in biennial and annual., 2019, 9: 14911.
[19] Ponjavic J, Ponting C P, Lunter G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs., 2007, 17: 556–565.
[20] Swiezewski S, Liu F Q, Magusin A, Dean C. Cold-induced silencing by long antisense transcripts of anPolycomb target., 2009, 462: 799–802.
[21] Song J H, Cao J S, Yu X L, Xiang X. BcMF11, a putative pollen-specific non-coding RNA fromssp. chinensis., 2007, 164: 1097–1000.
[22] Song J H, Cao J S, Wang C G. BcMF11, a novel non-coding RNA gene from, is required for pollen development and male fertility., 2013, 32: 21–30.
[23] Joshi R K, Megha S, Basu U, Rahman M H, Kav N N V. Genome wide identification and functional prediction of long non-coding RNAs responsive toinfection in., 2016, 11: e0158784.
[24] Zhang J F, Wei L J, Jiang J, Mason A S, Li H J, Cui C, Chai L, Zheng B C, Zhu Y Q, Xia Q, Jiang L C, Fu D H. Genome-wide identification, putative functionality and interactions between lncRNAs and miRNAs inspecies., 2018, 8: 4960.
[25] Shen E H, Zhu X T, Hua S J, Chen H Y, Ye C Y, Zhou L H, Liu Q, Zhu Q H, Fan L J, Chen X. Genome-wide identification of oil biosynthesis-related long non-coding RNAs in allopolyploid., 2018, 19: 745.
[26] Tan X Y, Li S, Hu L Y, Zhang C L. Genome-wide analysis of long non-coding RNAs (lncRNAs) in two contrasting rapeseed (L.) genotypes subjected to drought stress and re-watering., 2020, 20: 81
[27] Rousseau-Gueutin M, Belser C, Silva C D, Richard G, Istace B, Cruaud C, Falentin C, Boideau F, Boutte J, Delourme R, Deniot G, Engelen S, de Carvalho J F, Lemainque A, Maillet L, Morice J, Wincker P, Denoeud F, Chèvre A M, Aury J M. Long-read assembly of thereference genome Darmor-bzh., 2020, 9: giaa137.
[28] Brown J, Pirrung M, McCue L A. FQC Dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool., 2017, 33: 3137–3139.
[29] Bolger A M, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data., 2014, 30: 2114–2120.
[30] Kim D, Paggi J M, Park C, Bennett C, Salzberg S L. Graph-based genome alignment and genotyping with HISAT2 and HISAT- genotype., 2019, 37: 907–915.
[31] Pertea M, Pertea G M, Antonescu C M, Chang T C, Mendell J T, Salzberg S L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads., 2015, 33: 290–295.
[32] Love M I, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2., 2014, 15: 550.
[33] Pertea G, Pertea M. GFF Utilities: GffRead and GffCompare., 2020, 9: ISCB Comm J-304.
[34] Kang Y J, Yang D C, Kong L, Hou M, Meng Y Q, Wei L P, Gao G. CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features., 2017, 45: W12–W16.
[35] Wang L, Park H J, Dasari S, Wang S Q, Kocher J P, Li W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model., 2013, 41: e74.
[36] Li A, Zhang J Y, Zhou Z Y. PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme., 2014, 15: 311.
[37] Szcześniak M W, Rosikiewicz W, Makałowska I. CANTATAdb: a collection of plant long non-coding RNAs., 2016, 57: e8.
[38] Havlickova L, He Z S, Wang L H, Langer S, Harper A L, Kaur H, Broadley M R, Gegas V, Bancroft I. Validation of an updated associative transcriptomics platform for the polyploid crop speciesby dissection of the genetic architecture of erucic acid and tocopherol isoform variation in seeds., 2018, 93: 181–192.
[39] Raboanatahiry N, Chao H B, Dalin H, Pu S, Yan W, Yu L J, Wang B S, Li M T. QTL alignment for seed yield and yield related traits in., 2018, 9: 1127
[40] 王艳花, 谢玲, 杨博, 曹艳茹, 李加纳. 甘蓝型油菜开花相关基因的鉴定及进化与表达分析. 作物学报, 2019, 45: 1137–1145.
Wang Y H, Xie L, Yang B, Cao Y R, Li J N. Flowering genes in oilseed rape: identification, characterization, evolutionary and expression analysis., 2019, 45: 1137–1145 (in Chinese with English abstract).
[41] Qüesta J I, Song J, Geraldo N, An H H, Dean C.transcriptional repressor VAL1 triggers Polycomb silencing atduring vernalization., 2016, 353: 485–488.
[42] Blümel M, Dally N, Jung C. Flowering time regulation in crop: what did we learn from?, 2015, 32: 121–129.
[43] Teramoto H, Toyama T, Takeba G, Tsuji H. Noncoding RNA for, a cytokinin-repressed gene of cucumber., 1996, 32: 797–808.
[44] Li X R, Zhang S F, Bai J J, He Y K. Tuning growth cycles ofcropsnatural antisense transcripts of., 2016, 14: 905–914.
[45] Thalhammer A, Bryant G, Sulpice R, Hincha D K. Disordered cold regulated15 proteins protect chloroplast membranes during freezing through binding and folding, but do not stabilize chloroplast enzymes., 2014, 166: 190–201.
[46] Kidokoro S, Yoneda K, Takasaki H, Takahashi F, Shinozaki K, Yamaguchi-Shinozaki K. Different cold-signaling pathways function in the responses to rapid and gradual decreases in temperature., 2017, 29: 760–774.
[47] Hayami N, Sakai Y, Kimura M, Saito T, Tokizawa M, Iuchi S, Kurihara Y, Matsui M, Nomoto M, Tada Y, Yamamoto Y Y. The responses ofEarly Light-Induced Protein2 to ultraviolet B, high light, and cold stress are regulated by a transcriptional regulatory unit composed of two elements., 2015, 169: 840–855.
[48] Li X Y, Zhang G F, Liang Y H, Hu L, Zhu B N, Qi D M, Cui S J, Zhao H T.interacts with Nuclear Factor-Ys to promote flowering by directly regulatingin., 2021, 108: 1493–1506.
[49] Tang S, Zhao H, Lu S P, Yu L Q, Zhang G F, Zhang Y T, Yang Q Y, Zhou Y M, Wang X M, Ma W, Xie W B, Guo L. Genome- and transcriptome-wide association studies provide insights into the genetic basis of natural variation of seed oil content in., 2021, 14: 470–487.
[50] Basu U, Hegde V S, Daware A, Jha U C, Parida S K. Transcriptome landscape of early inflorescence developmental stages identifies key flowering time regulators in chickpea., 2022, 108: 565–583.
[51] Li Z W, Tian P, Huang T B, Huang J Z. Noncoding-RNA- mediated regulation in response to macronutrient stress in plants., 2021, 22: 11205.
[52] Fukuda M, Fujiwara T, Nishida S. Roles of non-coding RNAs in response to nitrogen availability in plants., 2020, 21: 8508.
[53] Zhou X X, Cui J, Meng J, Luan Y S. Interactions and links among the noncoding RNAs in plants under stresses., 2020, 133: 3235–3248.
[54] Song L, Fang Y, Chen L, Wang J, Chen X W. Role of non- coding RNAs in plant immunity., 2021, 2: 100180.
附表1 差异表达开花基因
Table S1 Differentially expressed genes related flowering time

拟南芥基因Arabidopsis thaliana gene-ID基因缩写Symbol甘蓝型油菜基因IDBrassica napus gene-ID开花调控途径Flowering regulation pathway AT4G11880AGL14A09p27210.1_BnaDAR其他途径 Other AT5G13790AGL15C09p63310.1_BnaDAR开花整合基因 Floral integrator AT1G69120AP1A02p15620.1_BnaDAR花分化和发育途径 Floral meristems and development AT1G69120AP1C02p17480.1_BnaDAR花分化和发育途径 Floral meristems and development AT3G21320AT3G21320A03p42190.1_BnaDAR其他途径 Other AT5G37780CAM1C09p30310.1_BnaDAR其他途径 Other AT4G17640CKB2C01p11310.1_BnaDAR光信号途径 Light singnalling AT2G42530COR15bA03p23680.1_BnaDAR春化途径 Vernalization AT2G42530COR15bA03p23700.1_BnaDAR春化途径 Vernalization AT2G42530COR15bC03p28240.1_BnaDAR春化途径 Vernalization AT2G42530COR15bC03p28260.1_BnaDAR春化途径 Vernalization AT1G12610DDF1A06p09040.1_BnaDAR赤霉素途径 Gibberellin AT1G12610DDF1A08p32730.1_BnaDAR赤霉素途径 Gibberellin AT1G12610DDF1C05p10640.1_BnaDAR赤霉素途径 Gibberellin AT1G12610DDF1C08p15540.1_BnaDAR赤霉素途径 Gibberellin AT1G12610DDF1C08p47250.1_BnaDAR赤霉素途径 Gibberellin AT5G54510DFL1A02p10230.1_BnaDAR其他途径 Other AT4G14690ELIP2A01p22980.1_BnaDAR光信号途径 Light singnalling
(续附表1)

拟南芥基因Arabidopsis thaliana gene-ID基因缩写Symbol甘蓝型油菜基因IDBrassica napus gene-ID开花调控途径Flowering regulation pathway AT4G14690ELIP2C01p28420.1_BnaDAR光信号途径 Light singnalling AT5G10140FLCA02p00340.1_BnaDAR春化途径 Vernalization AT5G10140FLCA03p16730.1_BnaDAR春化途径 Vernalization AT5G10140FLCC03p04920.1_BnaDAR春化途径 Vernalization AT5G10140FLCC09p67350.1_BnaDAR春化途径 Vernalization AT5G10140FLCC09p67380.1_BnaDAR春化途径 Vernalization AT1G30040GA2OX2C03p76330.1_BnaDAR赤霉素途径 Gibberellin AT5G67100ICU2A02p33280.1_BnaDAR花分化和发育途径 Floral meristems and development AT5G15970KIN2A02p03320.1_BnaDAR春化途径 Vernalization AT5G15970KIN2C02p08090.1_BnaDAR春化途径 Vernalization AT4G32040KNAT5C01p06890.1_BnaDAR赤霉素途径 Gibberellin AT5G65060MAF3A02p43650.1_BnaDAR春化途径 Vernalization AT5G65060MAF3C02p63700.1_BnaDAR春化途径 Vernalization AT3G01460MBD9C09p75140.1_BnaDAR光周期和生物钟途径Photoperiod and circadian clock AT3G62090PIL2C08p37050.1_BnaDAR光信号途径 Light singnalling AT1G13260RAV1A09p63190.1_BnaDAR光周期和生物钟途径Photoperiod and circadian clock AT2G03710SEPALLATA4A02p33530.1_BnaDAR花分化和发育途径 Floral meristems and development AT2G45660SOC1A05p05620.1_BnaDAR开花整合基因 Floral integrator AT2G45660SOC1C03p30160.1_BnaDAR开花整合基因 Floral integrator AT2G33810SPL3C04p15220.1_BnaDAR花分化和发育途径 Floral meristems and development AT3G16640TCTPC01p47690.1_BnaDAR花分化和发育途径 Floral meristems and development
附表2 差异表达LncRNA涉及到的开花基因
Table S2 Differentially expressed LncRNA related flowering time gene
拟南芥基因Arabidopsis thaliana gene-ID基因缩写Symbol甘蓝型油菜基因IDBrassica napus gene-ID开花调控途径Flowering regulation pathway AT4G14690ELIP2A01p22980.1_BnaDAR光信号途径 Light singnalling AT5G15970KIN2A02p03320.1_BnaDAR春化途径 Vernalization AT1G69120AP1A02p15620.1_BnaDAR花分化和发育途径 Floral meristems and development AT1G78440GA2OX1A02p22190.1_BnaDAR赤霉素途径 Gibberellin AT2G42530COR15bA03p23680.1_BnaDAR春化途径 Vernalization AT2G42530COR15bA03p23700.1_BnaDAR春化途径 Vernalization AT2G45660SOC1A03p25280.1_BnaDAR开花整合基因 Floral integrator AT2G45660SOC1A05p05620.1_BnaDAR开花整合基因 Floral integrator AT5G11260HY5A10p26460.1_BnaDAR光信号途径 Light singnalling AT5G15970KIN2C02p08090.1_BnaDAR春化途径 Vernalization AT1G69120AP1C02p17480.1_BnaDAR花分化和发育途径 Floral meristems and development AT4G25470CBF2C03p19680.1_BnaDAR春化途径 Vernalization AT2G42530COR15bC03p28240.1_BnaDAR春化途径 Vernalization AT2G42530COR15bC03p28260.1_BnaDAR春化途径 Vernalization AT1G12610DDF1C08p47250.1_BnaDAR赤霉素途径 Gibberellin AT5G20730NPH4C09p55450.1_BnaDAR光信号途径 Light singnalling
附表3 差异表达基因与差异表达LncRNA候选靶基因
Table S3 Differentially expressed genes and differentially expressed candidate target genes of LncRNA
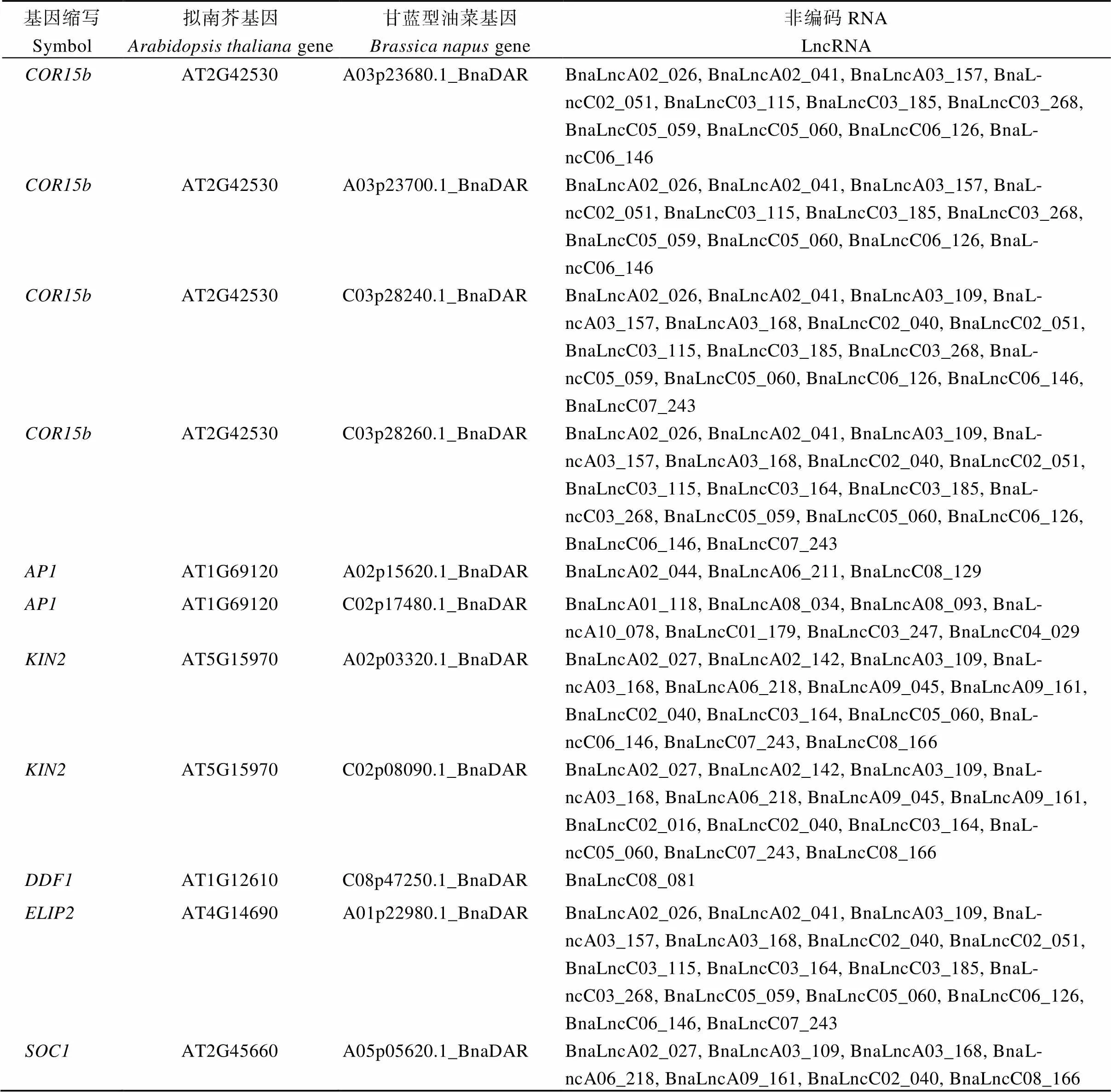
基因缩写Symbol拟南芥基因Arabidopsis thaliana gene甘蓝型油菜基因Brassica napus gene非编码RNALncRNA COR15bAT2G42530A03p23680.1_BnaDARBnaLncA02_026, BnaLncA02_041, BnaLncA03_157, BnaLncC02_051, BnaLncC03_115, BnaLncC03_185, BnaLncC03_268, BnaLncC05_059, BnaLncC05_060, BnaLncC06_126, BnaLncC06_146 COR15bAT2G42530A03p23700.1_BnaDARBnaLncA02_026, BnaLncA02_041, BnaLncA03_157, BnaLncC02_051, BnaLncC03_115, BnaLncC03_185, BnaLncC03_268, BnaLncC05_059, BnaLncC05_060, BnaLncC06_126, BnaLncC06_146 COR15bAT2G42530C03p28240.1_BnaDARBnaLncA02_026, BnaLncA02_041, BnaLncA03_109, BnaLncA03_157, BnaLncA03_168, BnaLncC02_040, BnaLncC02_051, BnaLncC03_115, BnaLncC03_185, BnaLncC03_268, BnaLncC05_059, BnaLncC05_060, BnaLncC06_126, BnaLncC06_146, BnaLncC07_243 COR15bAT2G42530C03p28260.1_BnaDARBnaLncA02_026, BnaLncA02_041, BnaLncA03_109, BnaLncA03_157, BnaLncA03_168, BnaLncC02_040, BnaLncC02_051, BnaLncC03_115, BnaLncC03_164, BnaLncC03_185, BnaLncC03_268, BnaLncC05_059, BnaLncC05_060, BnaLncC06_126, BnaLncC06_146, BnaLncC07_243 AP1AT1G69120A02p15620.1_BnaDARBnaLncA02_044, BnaLncA06_211, BnaLncC08_129 AP1AT1G69120C02p17480.1_BnaDARBnaLncA01_118, BnaLncA08_034, BnaLncA08_093, BnaLncA10_078, BnaLncC01_179, BnaLncC03_247, BnaLncC04_029 KIN2AT5G15970A02p03320.1_BnaDARBnaLncA02_027, BnaLncA02_142, BnaLncA03_109, BnaLncA03_168, BnaLncA06_218, BnaLncA09_045, BnaLncA09_161, BnaLncC02_040, BnaLncC03_164, BnaLncC05_060, BnaLncC06_146, BnaLncC07_243, BnaLncC08_166 KIN2AT5G15970C02p08090.1_BnaDARBnaLncA02_027, BnaLncA02_142, BnaLncA03_109, BnaLncA03_168, BnaLncA06_218, BnaLncA09_045, BnaLncA09_161, BnaLncC02_016, BnaLncC02_040, BnaLncC03_164, BnaLncC05_060, BnaLncC07_243, BnaLncC08_166 DDF1AT1G12610C08p47250.1_BnaDARBnaLncC08_081 ELIP2AT4G14690A01p22980.1_BnaDARBnaLncA02_026, BnaLncA02_041, BnaLncA03_109, BnaLncA03_157, BnaLncA03_168, BnaLncC02_040, BnaLncC02_051, BnaLncC03_115, BnaLncC03_164, BnaLncC03_185, BnaLncC03_268, BnaLncC05_059, BnaLncC05_060, BnaLncC06_126, BnaLncC06_146, BnaLncC07_243 SOC1AT2G45660A05p05620.1_BnaDARBnaLncA02_027, BnaLncA03_109, BnaLncA03_168, BnaLncA06_218, BnaLncA09_161, BnaLncC02_040, BnaLncC08_166
附表4 甘蓝型油菜收集的QTL信息汇总
Table S4 Summary of collected QTLs in

QTL性状 QTL trait数目Number 开花时间Flowering times181 千粒重Thousand seed weight160 株高Plant height124 种子产量Seed yield89 成熟时间Maturity time73 第一分支数Number of primary branches53 其他Others292 A基因组数目A genome numbers624 C基因组数目C genome numbers348 总计Total972
Preliminary exploration of the role of LncRNA in the ecotype differentiation ofL.
YANG Tai-Hua, YANG Fu-Quan, GAO Geng-Dong, YIN Shuai, JIN Qing-Dong, XU Lin-Shan, KUAI Jie, WANG Bo, XU Zheng-Hua, GE Xian-Hong, WANG Jing*, and ZHOU Guang-Sheng
College of Plant Science and Technology, Huazhong Agricultural University, Wuhan 430070, Hubei, China
L. is one of the important oil crops in China. Based on its different requirements for vernalization temperature during flower transition,can be divided into three ecotypes (spring, semi-winter, and winter ecotype). Previous studies found that long non-coding RNA (LncRNA) can regulate gene expression level in multiple levels and participate in the regulation of plant growth and development. In, LncRNA can regulate flowering time by regulating the expression of genes related to vernalization pathway. In this study, to preliminarily explore the ecotype differentiation and adaptive formation among the three ecotypes of, three different ecotypes ofwere used as the materials and high-throughput sequencing technology was used to sequence mRNA and LncRNA in seeding leaves. The GO and KEGG enrichment analysis of co-differential expression genes of three ecotypes ofshowed that there were a lot of differences in the anabolism of basic compounds, especially lipid compounds. A total of 3775 LncRNA sequences were identified from the three ecotypes of, among which 285 LncRNA were differentially expressed in two or three ecotype combinations, and 1517 candidate target genes were involved. The candidate target genes of these differential expression LncRNA were also enriched in large number of anabolism related pathways of basic compounds. Based on conjoint analysis of mRNA and LncRNA, we predicted a putative regulatory network in the flowering genes, included eight flowering time genes and 23 LncRNA, which were involved in the regulation of temperature and light signal pathways. By analyzing the location of QTLs for important agronomic traits in, we found that about 90% of LncRNA and QTLs intervals were overlapped. The distribution of different expression LncRNA and the location of QTLs were different in three ecotypes of, which suggesting that LncRNA played an important role in ecotype differentiation and formation of important agronomic traits in.
; ecotype; long non-coding RNA; flowering genes; QTLs
10.3724/SP.J.1006.2023.24105
本研究由国家重点研发计划项目“大田经济作物优质丰产的生理基础与调控”(2018YFD1000900)资助。
This study was supported by the National Key Research and Development Program of China “Physiological Basis and Agronomic Management for High-quality and High-yield of Field Cash Crops” (2018YFD1000900).
王晶, E-mail: wangjing@mail.hzau.edu.cn
E-mail: taihua1996@gmail.com
2022-04-27;
2022-09-05;
2022-09-15.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20220914.1812.007.html
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
