掺杂对GaN晶体力学性能影响的研究
2023-03-14王海笑李腾坤夏政辉陈科蓓张育民王鲁华高晓东任国强
王海笑,李腾坤,夏政辉,陈科蓓,张育民,3,王鲁华,高晓东,任国强,徐 科,3,4
(1.中国科学院苏州纳米技术与纳米仿生研究所,苏州 215123;2.中国科学技术大学纳米科学与技术学院,合肥 230026;3.苏州纳维科技有限公司,苏州 215123;4.江苏第三代半导体研究院,苏州 215000)
0 引 言

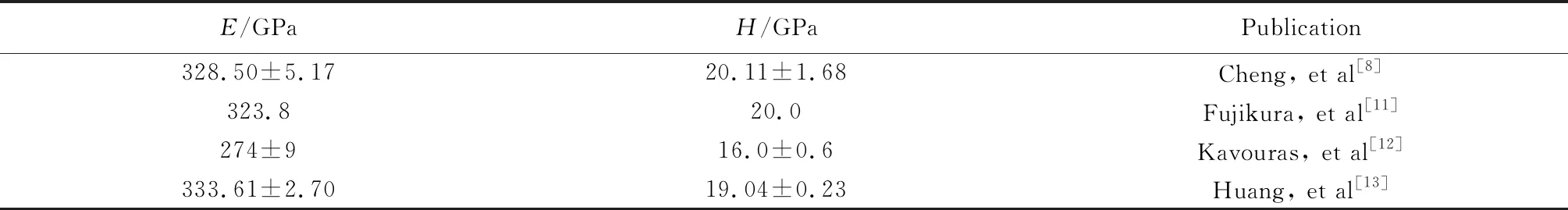
表1 研究人员测试的GaN单晶的弹性模量(E)和硬度(H)
影响GaN力学性能的主要因素有位错密度、残余应力、掺杂等。研究掺杂对GaN晶体力学性能的影响有助于解决其在工艺加工、器件制作及应用等过程中出现的裂纹等问题。本文利用纳米压痕技术实验方法,通过测试掺杂不同类型元素及重掺杂的GaN单晶的力学性能,研究了掺杂对GaN单晶力学性能特别是硬度的影响。
1 实 验
使用氢化物气相外延(hydride vapor phase epitaxy, HVPE)法制备了不同掺杂类型(非掺、Si掺、Fe掺)的GaN单晶,其中Si掺GaN掺杂浓度为1×1018cm-3(原子数分数,下同),Fe掺GaN掺杂浓度为5×1018cm-3。实验中采用的自支撑HVPE GaN样品厚度约为350 μm。HVPE GaN样品中O元素含量约为4×1016cm-3;非掺和Fe掺的HVPE GaN中Si元素的含量约为2×1017cm-3。氨热法的基本原理是:在准热力学平衡状态下,利用GaN多晶在氨中的溶解度随温度变化的原理,在温度梯度作用下,GaN从过饱和溶液中析出,最终在GaN籽晶上形核生长。设置生长温度为400~600 ℃,使生长压力达到200~300 MPa,生长时间为30 d以上,以HVPE GaN为籽晶,获得掺杂多种元素的氨热GaN单晶(ammonothermal GaN, Am-GaN)。
本文中纳米压痕测试的力学分析方法完全基于Oliver-Pharr法[14-15]。在纳米压痕实验中,选用Berkovich金刚石压头(曲率半径R=50 nm)测试样品的弹性模量和硬度,将压痕深度设置为500 nm,使用Keysight G200纳米压痕仪在每个样品上测试至少20个有效点,以减少数据误差。采用原子力显微镜(atomic force microscope, AFM)、透射电子显微镜(transmission electron microscope, TEM)、扫描电子显微镜(scanning electron microscope, SEM)和阴极荧光光谱(cathodoluminescene, CL)对样品的压痕形貌和位错滑移特性进行表征。在研究Am-GaN的位错滑移特点时,为了保证滑移带的对称性,采用尖端直径为5 μm的圆锥压头进行纳米压痕实验。在制备TEM样品时,用聚焦离子束(focused ion beam, FIB)在距离样品表面约150 nm处切下一个切片层,并且从样品表面的压痕中心处切下一个截面层。
采用二次离子质谱法(secondary ion mass spectroscopy, SIMS)测定了样品中掺杂元素的含量。利用高分辨率X射线衍射(high resolution X-ray diffraction, HR-XRD)仪(Bruker D8 Discover,狭缝的选择为1 mm×10 mm)测定样品的摇摆曲线半峰全宽(full width at half maximum, FWHM),以表征GaN的晶体质量。文献[16]详细描述了HR-XRD测定晶体晶格常数的方法。
2 结果与讨论
采用HVPE法生长得到非掺GaN、Si掺GaN和Fe掺GaN。采用氨热法获得了重掺杂的氨热GaN单晶,H和O的浓度为1019~1020cm-3;Si、Fe、Mg的浓度为1018~1019cm-3。表2给出了实验中GaN样品的FWHM和弹性模量(E)、硬度(H)及其均方误差的值。HVPE GaN的(002)FWHM非常接近,说明三种GaN的结晶质量基本一致。Am-GaN的(002)FWHM约为94″,略大于本文使用的HVPE GaN样品的。四种试样的弹性模量平均值分别为309.9、307.6、313.1和312.1 GPa,硬度平均值分别为17.40、17.61,17.53和18.42 GPa。可以发现,Si掺、Fe掺的硬度都比非掺GaN更大,特别是重掺杂的氨热GaN相对非掺GaN硬度增大约1 GPa。

表2 样品的弹性模量(E)和硬度(H)及其均方误差
本文尝试从Oliver-Pharr的力学分析方法中研究造成这种现象的原因。事实上,纳米压痕实验中硬度的测试是基于公式(1):
H=Pmax/A
(1)
式中:Pmax为压痕载荷;A为接触面积(不是压痕本身的面积)。金属、石英等材料的力学性能测试中也会出现硬度数据不准确的情况,在残余应力的影响下,压痕周围可能会出现材料的堆积或下沉行为,从而导致接触面积的变化[17-20]。然而Per-Lennart的研究也表明,在纳米压痕试验中,残余应力与硬度之间没有关系[21]。为了排除这一因素的影响,如图1所示,用AFM表征了四类样品的压痕形貌。显然,对于非掺和掺杂GaN样品,其表面的压痕周围都没有堆积或下沉行为,说明接触面积变化引起的数据误差很小。同时,在压痕尖端没有裂纹产生,能够说明实验数据的准确性。
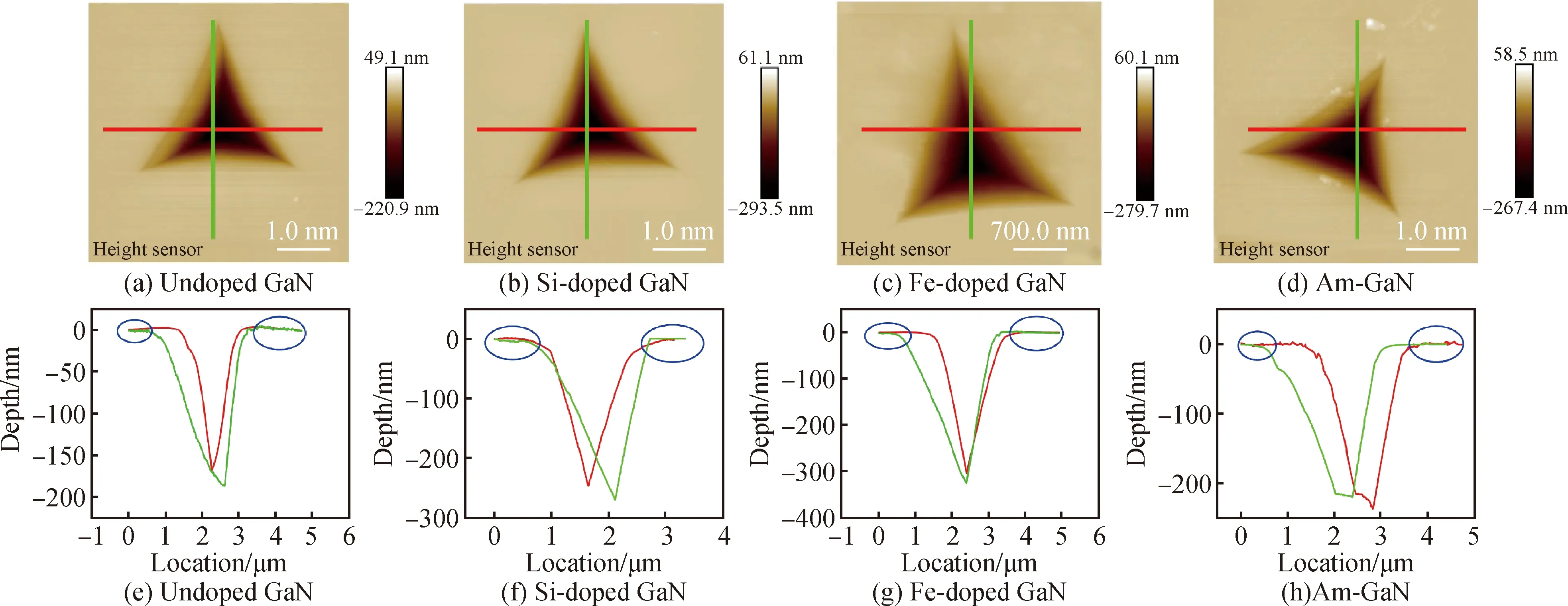
图1 非掺、Si掺、Fe掺和氨热GaN压痕的AFM照片(a)~(d)及其各自的压痕表面轮廓(e)~(h)
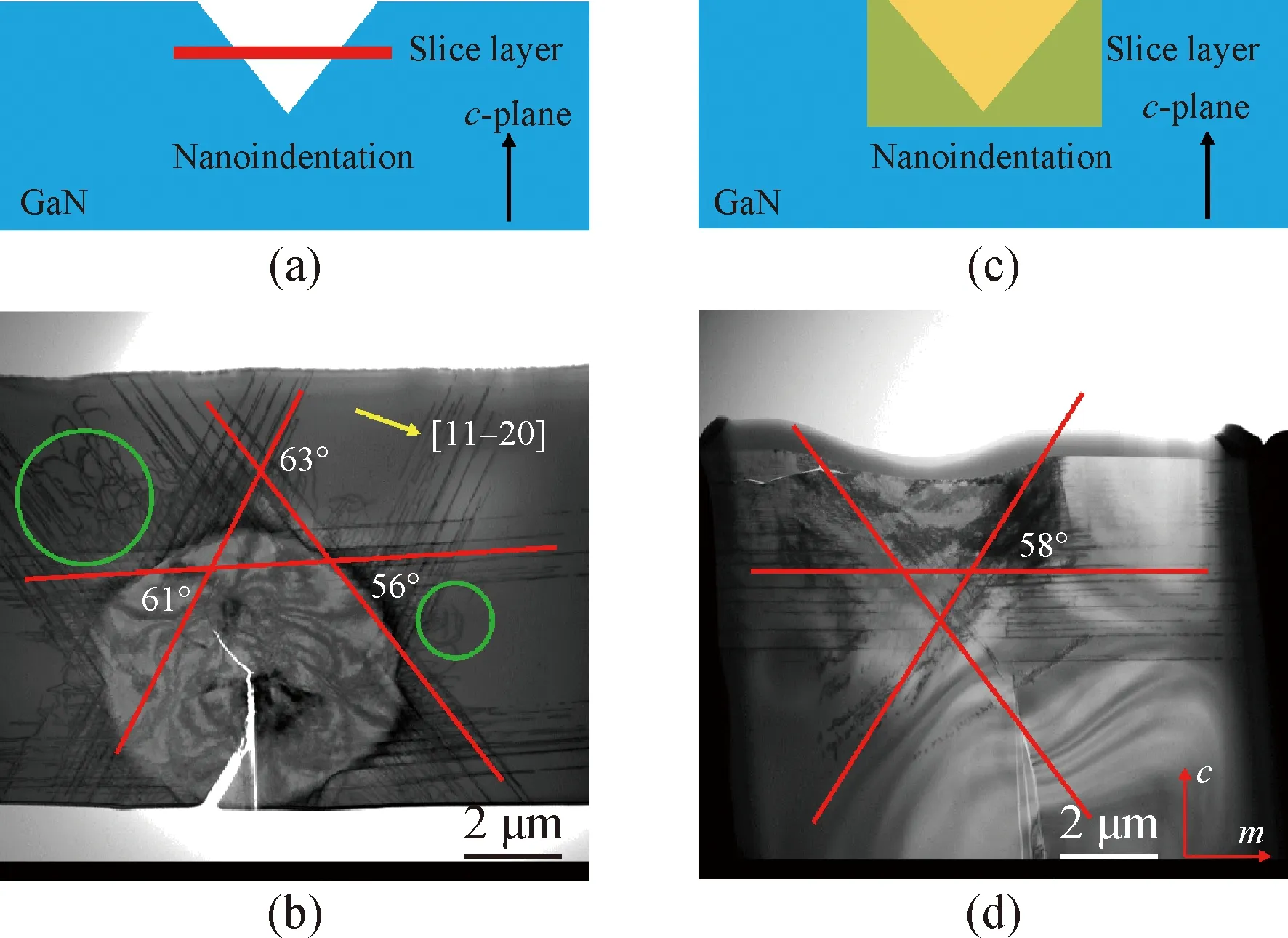
图2 Am-GaN纳米压痕结构的切片层示意图(a)及其TEM明场图像(b),截面切片层示意图(c)及其TEM明场图像(d)
虽然有研究者试图解释GaN在掺杂后硬度发生变化的原因,但在这个问题上并没有令人信服的结论。Fujikura等[24]推测GaN纳米压痕硬度变化的原因是生长条件引起的GaN晶体内氮空位浓度的变化。Evtimova等[25]对掺杂不同Si元素含量的GaN薄膜进行了显微硬度分析,认为硬度的变化可能与载流子浓度、迁移率以及一些受体缺陷和空位有关。
本文对GaN表面压痕产生的滑移带进行了表征。图3显示了非掺和Si掺GaN的残余压痕的CL照片,其中一个残余压痕的SEM照片被放置在图片右上角。结果表明,非掺GaN压痕的滑移带平均长度在95~100 μm。而对于Si掺的GaN,其滑移带平均长度仅为75~80 μm。Si掺GaN相对非掺样品滑移带长度缩短的原因可能是掺杂的杂质元素阻碍了晶体内的位错增殖和滑移。然而,对于Fe掺和氨热GaN,由于其载流子浓度很低,无法用CL观察到完整的滑移带。

图3 压痕室温全色CL照片
为了进一步揭示硬度变化的原因,通过HR-XRD测试了多个样品的晶格常数,如图4所示。在晶格常数的测试中,c是直接测量的,但a需要根据晶面夹角计算,误差较大,本文只给出c的值。对HVPE GaN来说,晶格常数变化相对较小,掺杂Si和Fe的GaN晶体,其晶格常数都有一定程度的增加,氨热GaN的晶格常数增大非常明显,这在很大程度上是由于多种元素掺杂的影响。结合不同掺杂类型GaN单晶的晶格常数和硬度数据,推测晶格常数的变化可能是引起GaN硬度的变化的主要原因之一,但具体的影响机制还需要进一步研究。
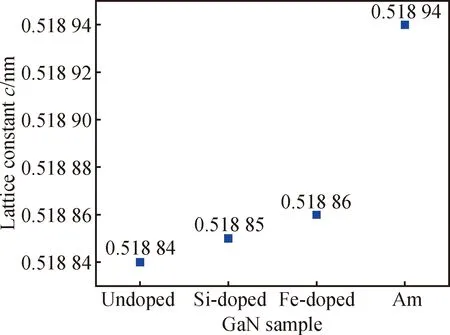
图4 HR-XRD测定不同掺杂类型GaN样品的晶格常数c
3 结 论
本文采用纳米压痕法测定了HVPE生长的不同掺杂类型(非掺、Si掺杂、Fe掺杂)GaN单晶的弹性模量和硬度,发现元素的掺杂会对GaN的硬度产生重要影响。实验结果表明,Si掺GaN和Fe掺GaN的硬度较非掺GaN有所提升。实验过程中,利用HRXRD、AFM和TEM表征排除了位错滑移、接触面积变化等因素对GaN硬度可能存在的影响。进一步研究了掺杂多种杂质元素的氨热GaN单晶的力学性能,氨热法生长的重掺杂GaN相对于非掺GaN,硬度提升约1 GPa。对GaN晶格常数和滑移带长度的测试结果表明,GaN硬度增大的主要原因是掺杂元素形成的缺陷阻碍了位错的增殖、滑移,以及掺杂元素引起的GaN晶格常数的变化。本研究为解决GaN单晶生长和加工过程中的开裂问题提供了新的思路,也为GaN基器件的制备和封装提供了力学性能方面的理论支持。
