近红外漫反射光谱法快速测定麦冬多指标成分含量
2023-03-11祁梅顾志荣李芹王安红葛斌
祁梅 ,顾志荣, ,李芹 ,王安红 ,葛斌
1.甘肃省人民医院,甘肃 兰州 730000; 2.甘肃省中医药研究中心,甘肃 兰州 730000;3.西南医科大学附属医院,四川 泸州 646000
麦冬为百合科沿阶草属植物麦冬Ophiopogon japonicus(L. f.)Ker-Gawl.的干燥块根,被《神农本草经》列为上品[1],主要产于四川三台和浙江杭州一带,分别称为“川麦冬”和“浙麦冬”,其中浙麦冬品质较川麦冬为优,但生产周期较川麦冬长,种植成本高,且近年生产规模有所萎缩[2]。二者属于不同产地的同一种属来源的中药,其化学成分及药理作用相同。研究表明,麦冬含有皂苷类、黄酮类、多糖类等有效成分[3],具有抗动脉粥样硬化[4]、抗心肌缺血[5]、抗肿瘤[6]、抗炎[7]、抗氧化[8]、增强免疫[9]、镇咳[10]、降血糖[11]等广泛的作用。
2020年版《中华人民共和国药典》麦冬项下仅以总皂苷含量进行质量控制,未将其他有效成分、有效部位或指标性成分作为质控指标[12],因此难以对麦冬进行快速、全面的质量控制与评价。目前已实现麦冬中总皂苷、总黄酮、总多糖的近红外光谱(NIR)快速测定[13-14],对完善麦冬的质量控制体系作出了技术示范,但主要指标性成分及单体有效成分的快速测定对麦冬的质量控制更具有实际价值,而鲜见报道。本研究采集不同产区、不同采收期的麦冬样品,建立其代表性成分麦冬皂苷B、麦冬皂苷D、麦冬皂苷D′、甲基麦冬二氢高异黄酮A、甲基麦冬二氢高异黄酮B含量的NIR定量分析模型,实现其含量的快速测定,为麦冬的快速、全面质量控制与评价提供参考。
1 仪器与试药
Nicolet-6700型傅里叶变换近红外光谱仪(美国Thermo公司),Agilent 1200型高效液相色谱仪(美国Agilent公司),Alltech 3300型蒸发光散射检测器(ELSD,美国Grace公司),Milli-Q Advantage A10超纯水仪(法国Merck Millipore公司),DZF-6090型真空干燥箱(上海一恒科学仪器有限公司),AL204型万分之一电子天平(瑞士Mettler-Toledo公司),DD-5M型离心机(湘仪离心机仪器有限公司),SB25-12DTD型超声波清洗机(宁波新芝生物科技股份有限公司)。
120批麦冬样品,采集(购买)于四川、浙江的3个主要产区,见表1。所有样品均经甘肃中医药大学药学院中药鉴定教研室李硕副教授鉴定为百合科沿阶草属植物麦冬Ophiopogon japonicus(L. f.)Ker-Gawl.的干燥块根[12]。将样品粉碎至过80目筛,冷藏备用。麦冬皂苷B对照品(批号CHB180122)、麦冬皂苷D对照品(批号CHB180120)、麦冬皂苷D′对照品(批号CHB180121)、甲基麦冬二氢高异黄酮A对照品(批号CHB190119)、甲基麦冬二氢高异黄酮B对照品(批号CHB190125),成都克洛玛生物科技有限公司,质量分数均不低于98%;甲醇、乙腈均为色谱纯,天津大茂化学试剂厂;其他试剂均为分析纯。
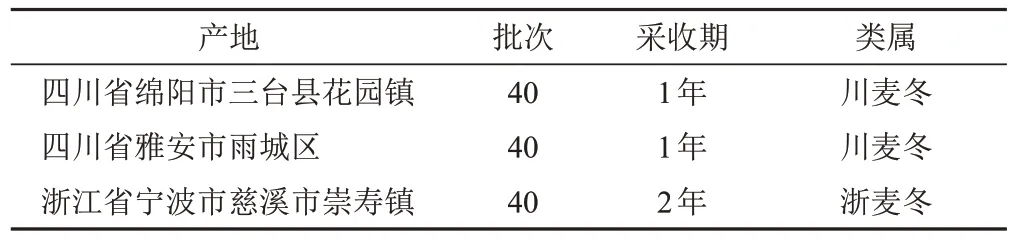
表1 麦冬样品来源信息
2 方法与结果
2.1 近红外光谱采集
仪器预热30 min后,采用积分球漫反射方式采集NIR。取于60 ℃干燥至恒重的麦冬粉末适量,混匀,置于样品旋转杯内,压实,以内置背景为参比,扫描范围10 000~4 000 cm-1,扫描64次,分辨率8 cm-1,温度(25±0.5)℃,相对湿度20%~30%,每批样品重复采集3次,取平均光谱[15]。120批麦冬样品NIR叠加图见图1。
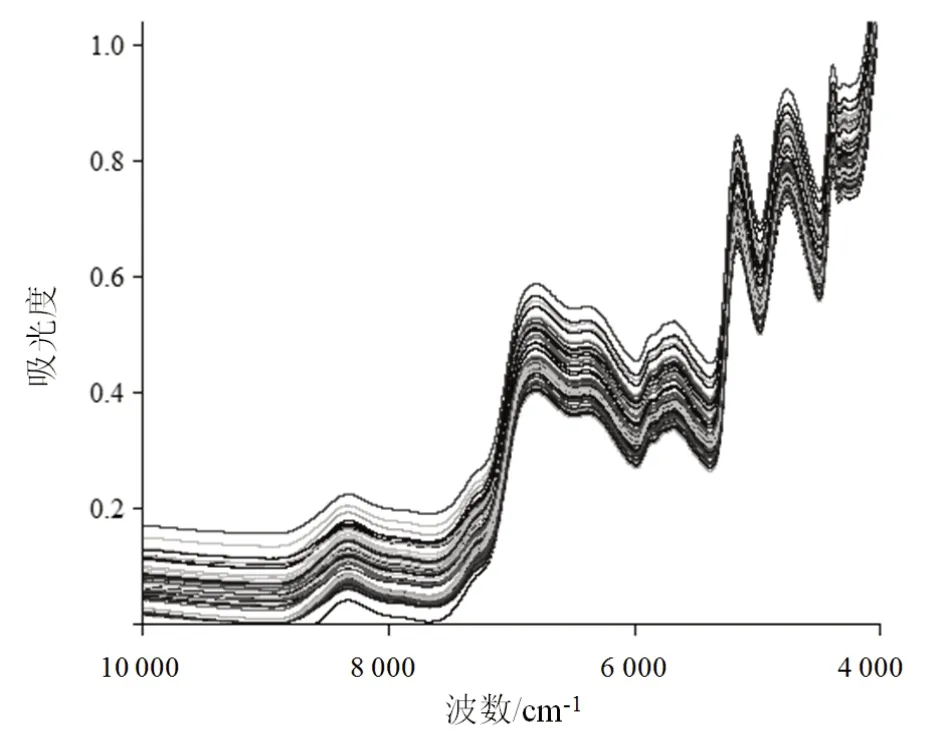
图1 120批麦冬样品NIR叠加图
2.2 化学参考值测定
2.2.1 供试品溶液制备
精密称取麦冬粉末约4.0 g,置于具塞三角瓶中,精密加入甲醇溶液50 mL,超声(频率40 kHz,功率500 W)提取60 min,过滤,滤液60 ℃水浴蒸至近干,转移至5 mL量瓶中,用甲醇洗涤并定容至刻度线,0.45 μm微孔滤膜过滤,取续滤液,即得。
2.2.2 混合对照品溶液制备
分别精密称取麦冬皂苷D、麦冬皂苷D′、麦冬皂苷B、甲基麦冬二氢高异黄酮A、甲基麦冬二氢高异黄酮B 7.72、8.62、5.51、10.14、7.53 mg,置于25 mL量瓶中,加甲醇适量,超声使完全溶解,并以甲醇定容至25 mL,制成浓度分别为0.308 8、0.344 8、0.220 4、0.405 6、0.301 2 mg/mL的混合对照品溶液。
2.2.3 色谱条件
检测器为ELSD,采用Agilent Extend C18色谱柱(4.6 mm×250 mm,5 μm),以乙腈(A)-水(B)为流动相,梯度洗脱(0~60 min,35%A → 65%A),流速1 mL/min,柱温35 ℃,漂移管温度100 ℃,氮气流速3.0 L/min,进样量20 μL。色谱图见图2。
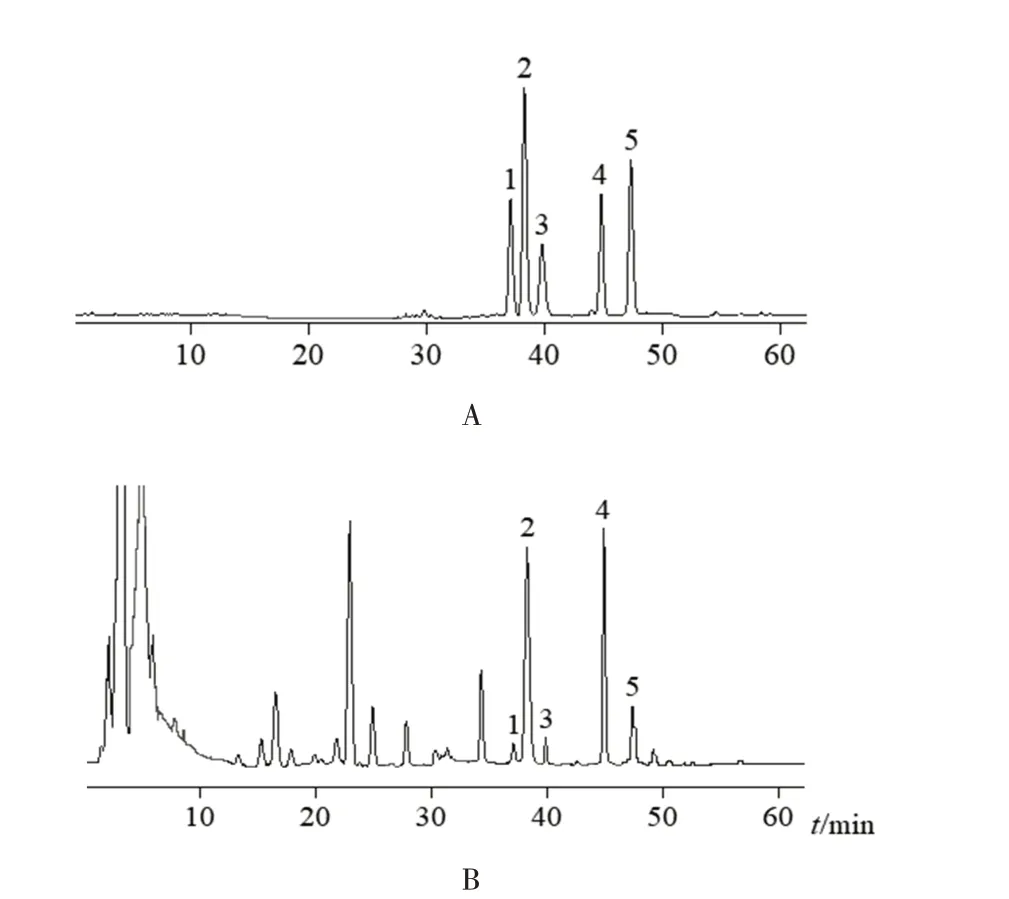
图2 麦冬中5种成分HPLC图
2.2.4 方法学考察及含量测定
经方法学考察,5种成分线性回归方程相关系数为0.998 8~0.999 5;精密度试验结果表明,5种成分峰面积RSD为0.71%~0.93%;稳定性试验结果表明,5种成分的溶液在24 h内稳定;重复性试验结果表明,5种成分峰面积RSD为1.45%~1.85%;5种成分平均加样回收率为98.93%~102.33%,RSD为1.76%~2.21%。表明本试验所采用的仪器及方法误差均符合要求。按“2.2.1”项下方法制备供试品溶液,按“2.2.3”项下色谱条件测定峰面积,采用外标标准曲线法计算样品中5种成分含量的化学参考值。
2.3 定量校正模型建立
2.3.1 异常样品剔除
异常样品指光谱数据存在较大误差的样品,这些样品会对所建模型的准确性及稳健性产生不利影响。本试验利用Dixon检验剔除异常光谱[16],结果共剔除8个光谱异常的样品。
2.3.2 样本集划分
将经过Dixon检验的112批样品按含量数值从小到大排列后,选取2/3作为校正集(75批),剩余1/3作为验证集(37批),并且确保验证集样品含量在校正集样品的含量范围之内[17]。样本集划分与含量分布结果见表2。
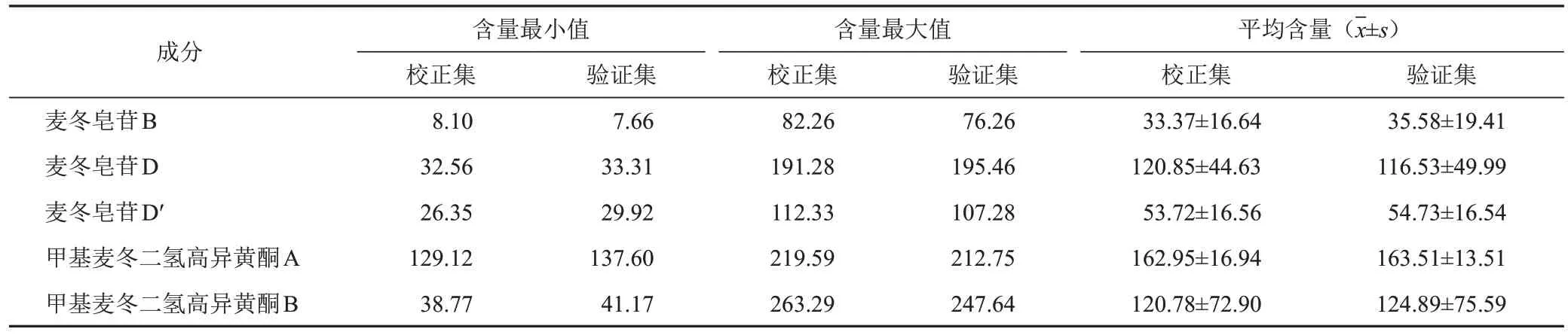
表2 5种成分样本集划分与含量分布结果(mg/g,n=3)
2.3.3 光谱预处理方法选择
NIR易受噪声、基线漂移、样品颗粒不均匀、光散射等多种因素干扰,引起随机误差,降低信噪比,使模型的准确性与稳健性降低,因此对光谱进行预处理非常重要。本试验考察未处理光谱(None)、标准正态变量校正(SNV)、多元散射校正(MSC)、一阶导数(FD)、二阶导数(SD)、11点移动窗口最小二乘多项式平滑(Savitzky-Golay smoothing,SG)和Norris导数滤波平滑等常见光谱预处理方法及组合方法。以决定系数(R2)、校正均方差(RMSEC)、预测均方差(RMSEP)和留一法交叉验证均方差(RMSECV)为指标,筛选不同建模方法。R2越接近1,RMSEC、RMSEP和RMSECV越接近0,模型的稳健性及预测效果越好。结果表明,对麦冬皂苷B、麦冬皂苷D、麦冬皂苷D′、甲基麦冬二氢高异黄酮A、甲基麦冬二氢高异黄酮B分别采用SNV+SD+SG、MSC+FD+SG、MSC+SD+SG、MSC+FD+Norris、MSC+FD+ SG进行光谱预处理所得模型预测效果最佳,见表3。
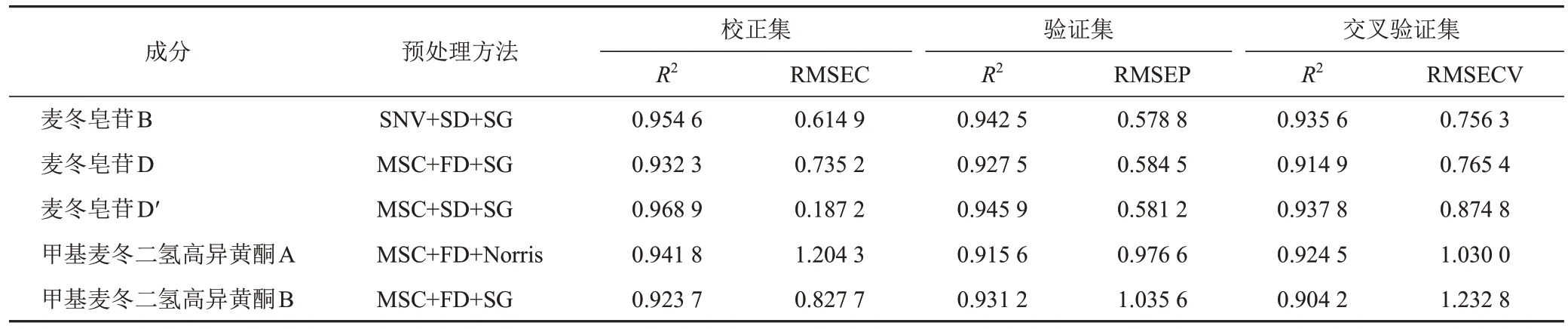
表3 5种成分最佳光谱预处理方法
2.3.4 光谱波段选择
采用筛选出的最佳光谱预处理方法处理光谱后,用TQ Analyst 8.0软件的自动优化功能,对不同光谱波段的建模效果进行考察,选取留一交叉验证R2最接近1的波段为最佳建模波段。可得麦冬皂苷B的最佳建模波段为8 105.4~7 316.6 cm-1及9 713.4~8 945.5 cm-1,麦冬皂苷D的最佳建模波段为9 554.6~7 098.4 cm-1,麦冬皂苷D′的最佳建模波段为8 253.1~6 765.2 cm-1、9 425.4~9 005.8 cm-1及9 637.1~9 458.9 cm-1,甲基麦冬二氢高异黄酮A的最佳建模波段为9 252.6~4 125.1 cm-1,甲基麦冬二氢高异黄酮B的最佳建模波段为9 134.2~4 254.2 cm-1。
2.3.5 因子数选择
采用内部交叉验证法考察因子数对RMSECV及交叉验证R2(%)的影响。当模型RMSECV较小且R2最接近1时,对应的因子数用于建模效果最佳。结果表明,麦冬皂苷B、麦冬皂苷D、麦冬皂苷D′、甲基麦冬二氢高异黄酮A、甲基麦冬二氢高异黄酮B的因子数分别选择10、9、10、6、5时建模效果最佳。
2.3.6 定量模型建立与评价
通过上述模型优化过程,得到麦冬中5种成分的PLS模型建立方法,见表4。校正模型的相关性见图3。其中,麦冬皂苷B定量校正模型R2=0.970 2,RMSEC=0.520 7,RMSEP=0.554 1,采用交叉验证法判断模型稳健性,交叉验证R2=0.943 0,RMSECV=0.646 5;麦冬皂苷D定量校正模型R2=0.949 8,RMSEC=0.662 7,RMSEP=0.315 7,交叉验证R2=0.920 6,RMSECV=0.358 3;麦冬皂苷D′定量校正模型R2=0.980 4,RMSEC=0.085 3,RMSEP=0.415 5,交叉验证R2=0.951 6,RMSECV=0.693 3;甲基麦冬二氢高异黄酮A定量校正模型R2=0.959 3,RMSEC=0.812 2,RMSEP=0.567 4,交叉验证R2=0.939 5,RMSECV=0.744 4;甲基麦冬二氢高异黄酮B定量校正模型R2=0.940 8,RMSEC=0.645 1,RMSEP=0.747 6,交叉验证R2=0.928 3,RMSECV=0.884 9。由图3可知,校正集与验证集样品均匀分布在回归线两侧,表明模型预测值与实测值(化学参考值)之间具有较好的相关性,模型预测性能较为理想。
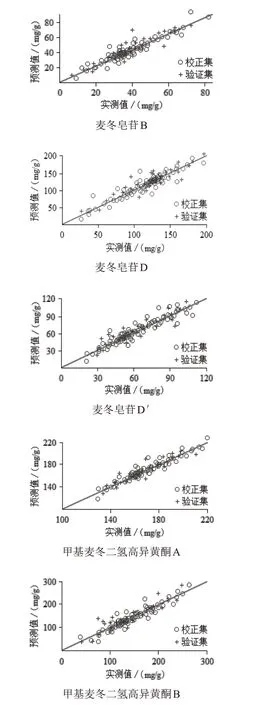
图3 5种成分参考值与模型预测值的相关性

表4 5种成分PLS模型建立方法
2.3.7 外部验证
选择15批样品扫描NIR,输入模型进行外部验证,检验模型的准确性,结果见表5~表9。可见,模型预测值的分布范围与实测值基本相符,相对误差均较小,预测结果较为准确。对各成分的模型预测值与实测值进行配对t检验,结果P值均大于0.05(α=0.05),预测值与实测值比较差异无统计学意义,表明2种检测方法之间的系统误差较小,可忽略不计。因此,利用NIR技术快速测定麦冬5种成分的含量是可行的。
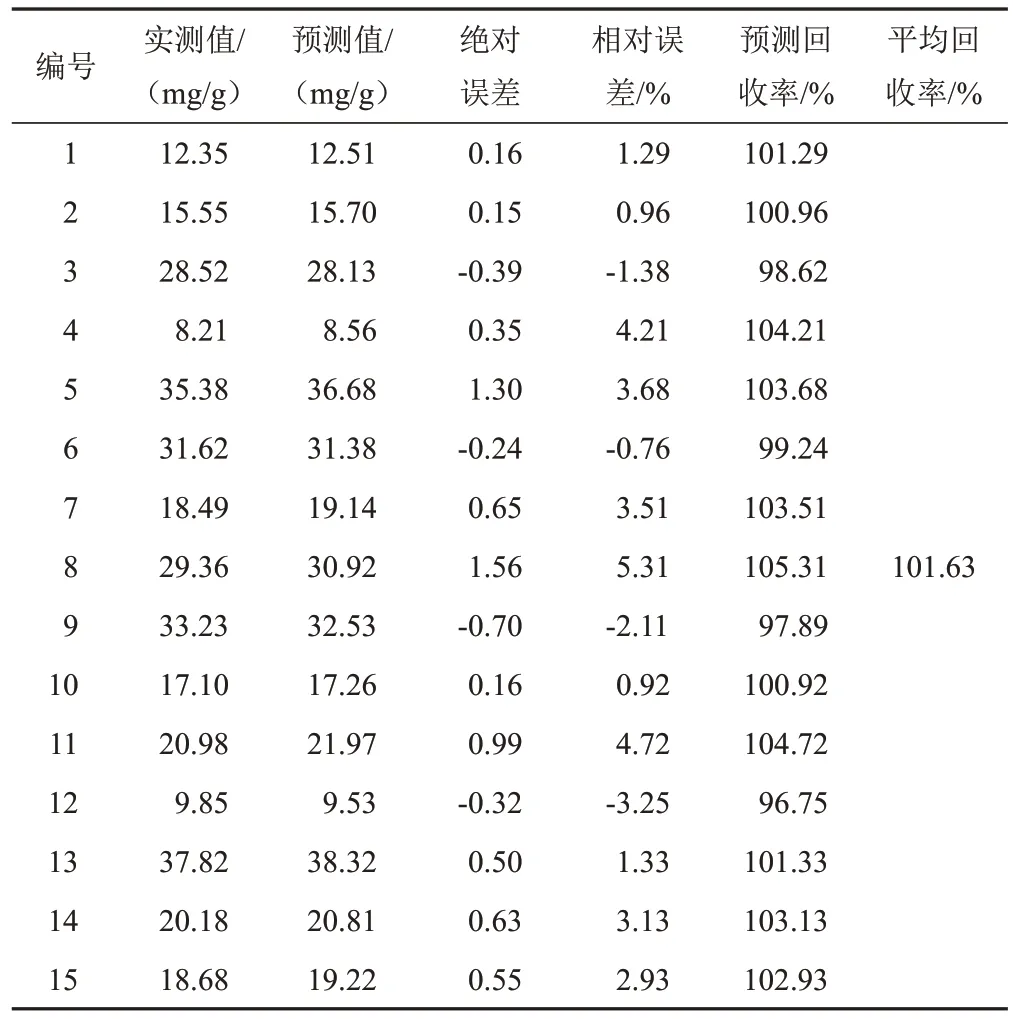
表5 麦冬皂苷B的PLS模型外部验证结果
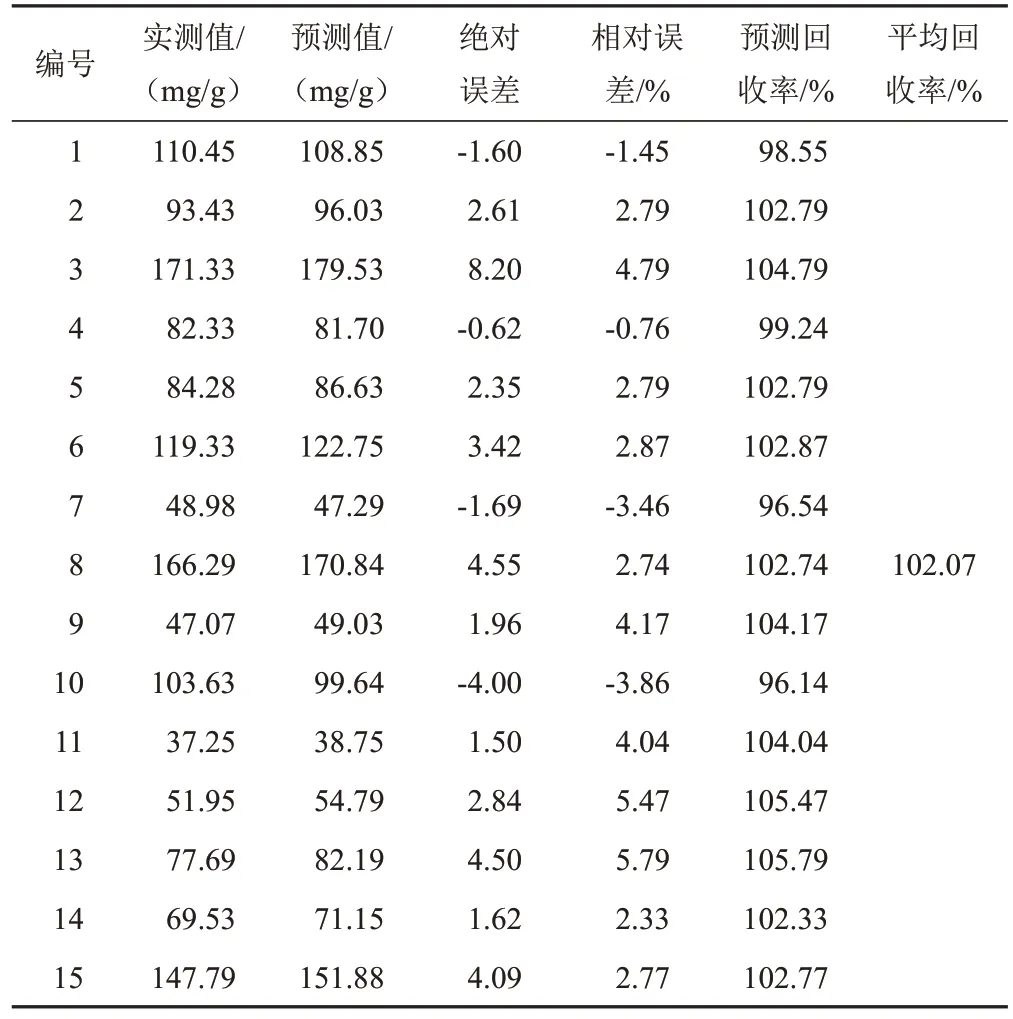
表6 麦冬皂苷D的PLS模型外部验证结果
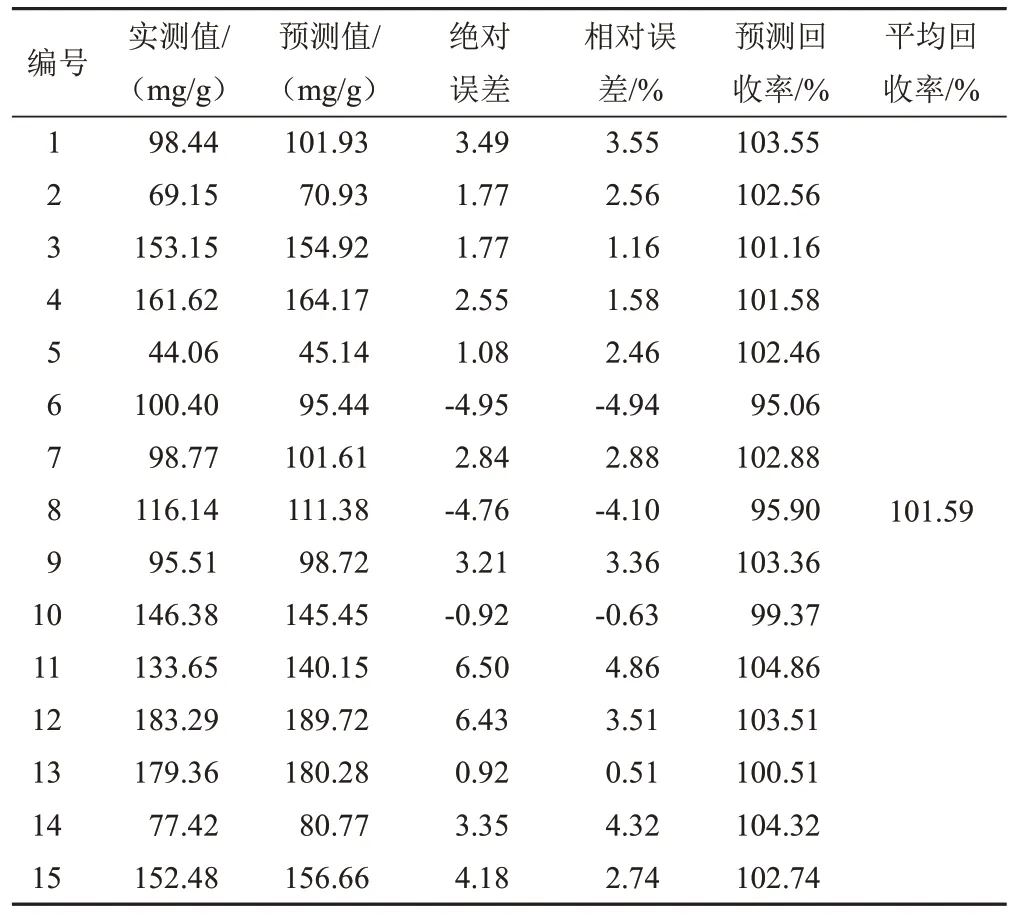
表9 甲基麦冬二氢高异黄酮B的PLS模型外部验证结果
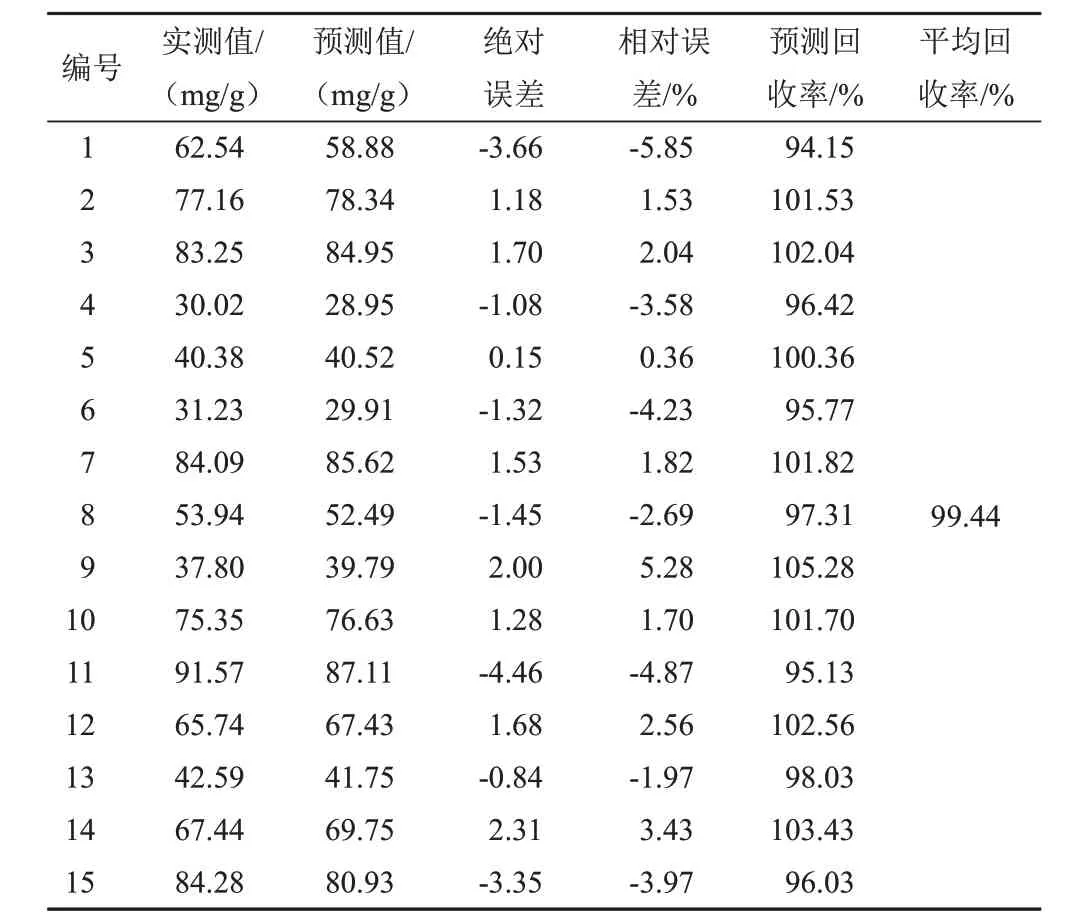
表7 麦冬皂苷D′的PLS模型外部验证结果
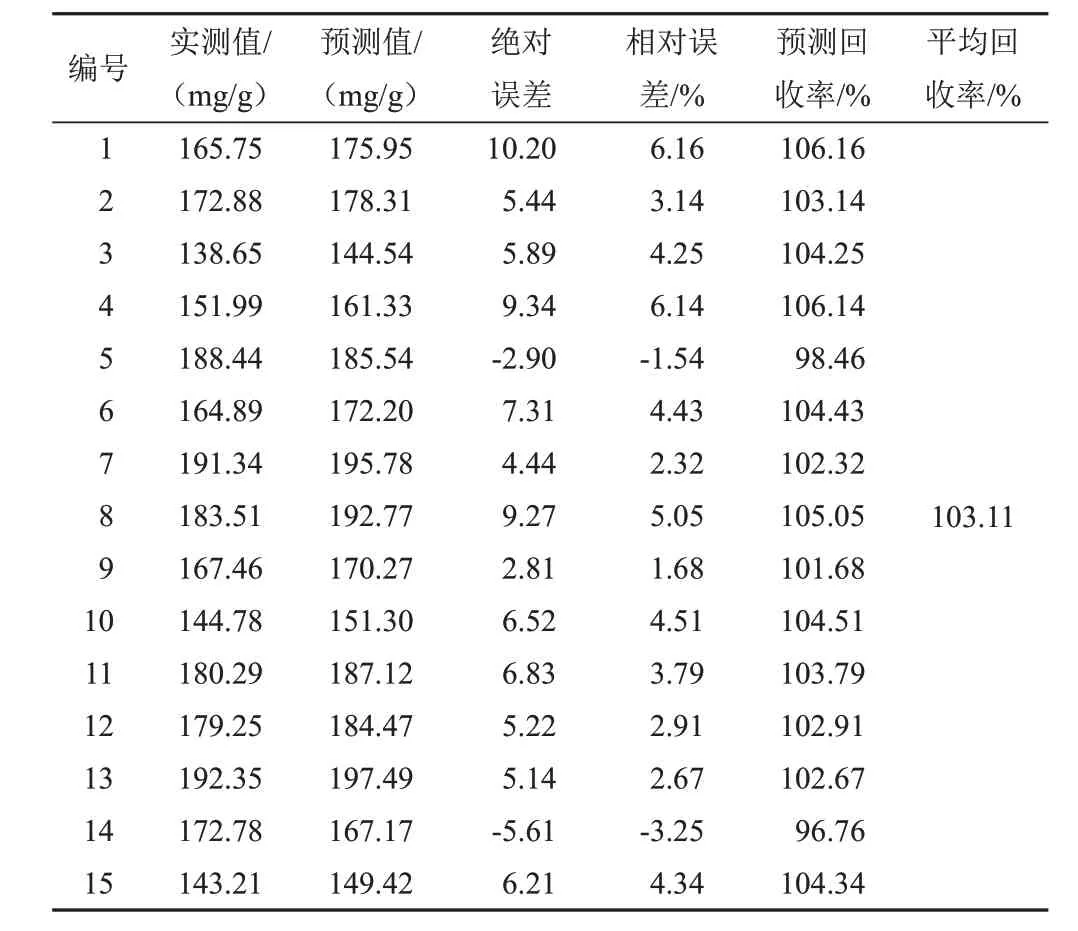
表8 甲基麦冬二氢高异黄酮A的PLS模型外部验证结果
3 讨论
本研究对象是百合科植物麦冬Ophiopogon japonicus(L. f)Ker-Gawl.,不包含山麦冬[即百合科植物湖北麦冬Liriope spicata(Thunb.)Lour. var.proliferaY. T. Ma和Liriope muscari(Decne.)Baily],2020年版《中华人民共和国药典》将麦冬与山麦冬严格区分为2种不同的中药。已有研究发现,山麦冬与麦冬的化学成分组成存在差异[18]。
传统的麦冬采收期多根据经验确定,如川麦冬在种植次年清明之后采收,而浙麦冬在种植后的第3年立夏后采收,两者种植区域的东西跨度较大,采收期时间跨度也较大,因此其生产、加工、流通、使用等各环节的质量控制较难掌握。2020年版《中华人民共和国药典》收载了麦冬水分、总灰分、水溶性浸出物和麦冬总皂苷的限量要求,规定含麦冬总皂苷以鲁斯可皂苷元计不得少于0.12%[12],但未对麦冬其他单体有效成分或有效部位进行要求。因此,目前尚缺少完善的含量标准评价麦冬质量及不同品种的差异。
川麦冬与浙麦冬化学成分种类及主要药理作用未见明显不同,但其成分含量存在明显差异,主要表现为浙麦冬中麦冬皂苷B、麦冬皂苷D′、甲基麦冬二氢高异黄酮A、甲基麦冬二氢高异黄酮B、总黄酮及总多糖的含量更高,因此浙麦冬质量更优[19]。麦冬总皂苷、总黄酮是麦冬的主要有效部位,是其抗动脉粥样硬化、抗炎、抗肿瘤的物质基础[4,20]。本研究建立的近红外光谱模型可以准确测试2个产地麦冬中的3种主要皂苷及2种主要黄酮的含量,模型预测结果与实测结果无明显差异,可为麦冬的质量控制提供快速、简便的分析方法及评价依据。
