A novel cerebrovascular drug-coated balloon catheter for treating symptomatic intracranial atherosclerotic stenosis lesions:Study protocol for a prospective, multicenter, single-arm, target-value clinical trial
2023-03-06QinhoDingWenboLiuJinggeZhoDehuGuoYoTngTengfeiZhouYnynHeFerinnHuiYonghongDingLingfuZhuZilngWngYingkunHeTinxioLi
Qinho Ding, Wenbo Liu, Jingge Zho, Dehu Guo, Yo Tng, Tengfei Zhou,Ynyn He, Ferinn K.Hui, Yonghong Ding, Lingfu Zhu, Zilng Wng,*,Yingkun He,***, Tinxio Li,**
a Department of Cerebrovascular Disease and Neurosurgery, Zhengzhou University People's Hospital, Henan University People's Hospital, Henan Provincial People's Hospital, Henan Provincial Neurointerventional Engineering Research Center, Henan International Joint Laboratory of Cerebrovascular Disease and Henan Engineering Research Center of Cerebrovascular Intervention, Zhengzhou, 450003, China
b Clinical Research Center, Zhengzhou University People's Hospital, Henan Provincial People's Hospital, Zhengzhou, 450003, China
c Neuroscience Institute, Queen's Medical Center, University of Hawaii, Honolulu, HI, USA
d Department of Radiology, Mayo Clinic, Rochester, MN, USA
Keywords:
ABSTRACT Background: Previous single-center studies have demonstrated that drug-coated balloons (DCBs) may reduce restenosis rates, which is an important factor affecting the prognosis for intracranial interventional therapy.However,currently available cardiac DCBs are not always suitable for the treatment of intracranial atherosclerotic stenosis(ICAS).This study aimed to evaluate the safety and efficacy of a novel DCB catheter designed for patients with severely symptomatic ICAS.
1.Introduction
Stroke is associated with the highest morbidity and mortality rates.1,2The 2014 Chinese Intracranial Atherosclerosis Study (CICAS) reported that the incidence of intracranial atherosclerotic stenosis (ICAS) in Chinese patients who experienced an ischemic stroke or transient ischemic attack (TIA) was 46.6 %.Patients with ICAS experience more severe symptoms and longer hospital stays, and the rate of stroke recurrence increases with the degree of stenosis.3Recent prospective, multi-center,randomized controlled trials have proved that aggressive drug therapy is the best treatment for patients with ICAS.4–6However, despite optimal medical therapy,symptoms and infarcts persist in some patients.
In addition to aggressive medical therapy and lifestyle management,the rapid advances and development of interventional therapies have provided new options for this high-risk patient subgroup.Percutaneous balloon angioplasty and stent implantation can quickly and effectively resolve regional brain hypoperfusion.Compared with balloon angioplasty,stenting has several disadvantages.First,the metal layer of a stent may increase the risk of thrombosis.Moreover, long-term dual antiplatelet therapy may increase the risk of bleeding.However, simple balloon angioplasty has a high restenosis rate,7which mitigates its effectiveness as monotherapy.
Drug-coated balloons (DCBs) that contain or are coated with lipophilic paclitaxel have also been developed.During balloon expansion,paclitaxel can irreversibly bind to cell microtubules8and penetrate and disperse into the vessel wall,9,10thus inhibiting endotheliosis and smooth muscle hyperplasia.This may reduce restenosis rates after balloon dilatation and improve patient prognosis.1,2,11However, currently used DCBs designed for cardiac indications are not always satisfactory for the treatment of ICAS because stiffer coronary drug balloon catheters cannot easily advance through tortuous cerebral vessels and intracranial stenoses, which may increase treatment failure rates.12Furthermore, intracranial balloon angioplasty is performed with a milder submaximal angioplasty, which requires the balloon to have appropriate characteristics.13To address these problems, we initiated a multicenter,prospective, single-aim, target-value clinical trial, which collected 155 cases from multiple centers from February 2021 to January 2022 to provide substantial evidence supporting the safety and effectiveness of a novel DCB catheter designed for patients with symptomatic,severe ICAS.
2.Method
2.1.Study device
The Ziyan device is a rapid-exchange DCB catheter that consists of 6 parts: a DCB, marker ring, distal rod, guidewire channel, proximal rod,and catheter adapter.The structure of the product is illustrated in Fig.1.The balloon is located at the end of the distal rod and can be expanded to a specific diameter under the recommended pressure.The 1.25 mm and 1.5 mm diameter balloons have a marker ring located in the middle of the balloon,and the other sizes of balloons have two marker rings located on the two shoulders of the balloon for intravascular visualization and localization.The coating drug–located on the outer surface of the balloon–is paclitaxel.The guidewire channel is located in the lumen of the distal rod and can accommodate a guidewire with a diameter not>0.014.
This DCB catheter was designed for ICAS dilation in intracranial arteries.After percutaneous puncture,lesions are initially predilated using a conventional balloon, which facilitates advancement and achieves immediate revascularization.The DCB catheter can then be placed across an appropriate intracranial artery lesion, and the balloon is inflated for 60 s to enable the drug on the surface of the balloon to transfer quickly and evenly to the lesion.The paclitaxel coating on the surface of the balloon can block the mitosis of vascular cells, thereby inhibiting excessive proliferation of the neointima and smooth muscle migration,and maintaining artery patency.

Fig.1.Schematic diagram of the balloon catheter section and balloon folding.
2.2.Study design
This was a prospective,multi-center,single-arm,target-value clinical trial initiated by 9 centers in China to evaluate the safety and efficacy of the Ziyan DCB catheter for the treatment of symptomatic ICAS.A total of 155 patients with intracranial atherosclerotic stenosis were recruited between February 2021 and January 2022.This study was approved by the Ethics Committee of Henan Provincial People's Hospital(Zhengzhou,China; reference number: 2020-145-03) and other research centers.A flow-diagram illustrating study flow is presented in Fig.2.
2.3.Participants
All patients fulfilled the inclusion criteria and none met any exclusion criteria.Eligible patients were enrolled in the study after obtaining informed consent.Details of the inclusion and exclusion criteria are listed in Tables 1 and 2,respectively.
2.4.Withdrawal criteria
During this clinical trial,subjects had the opportunity to withdraw at any time without reason.Patients who provided informed consent should inform the researchers immediately before choosing to discontinue the interventional treatment.Researchers had the discretion to suspend a trial based on their medical expertise.Patients were to discontinue treatment early if any of the following situations, but not limited to,occurred:

Table 1 Inclusion criteria.
1) If serious safety problems occurred during the trial, the safety of the subjects should be ensured in time,and the trial should be terminated if necessary.

Fig.2.Flow-diagram illustrating study flow.

Table 2 Exclusion criteria.
2) If major errors in the protocol were identified during the trial or there were major deviations, making it difficult to evaluate the safety and effectiveness of the trial product.
3) The sponsor requested suspension (for financial and management reasons).
4) The administrative authority cancelled the trial
If these situations were to occur, the sponsor was required to promptly report to the investigators of each research center,clinical trial institution, ethics committee, and the provincial food and drug administration where the sponsor is located.For subjects who were treated with the trial product,the sponsor must perform its responsibilities during the trial period in accordance with the laws and regulations of China and the content of informed consent.
2.5.Intervention technique
All patients were treated with dual antiplatelet drugs(aspirin 100 mg and clopidogrel 75 mg, once daily), and thromboelastography was performed.If drug resistance was observed,alternative drugs were selected(ticagrelor 90 mg, twice/day).Risk factors for atherosclerosis, such as blood pressure, blood glucose,and lipid levels,were addressed,and patients undergoing surgery were treated with dual antiplatelet drugs for at least 5 days before surgery.If the duration of dual antiplatelet therapy is less than 5 days,a loading dose of 300mg clopidogrel was administered.All surgeries were performed under general anesthesia, and the right femoral approach was performed using an 8 F sheath.Unfractionated heparin(70 U/kg)was administered intravenously in increments of 1000 U/h.Using a loach guidewire and angiographic catheter,the tip of the 6 F long sheath was placed at the level of the extracranial segment of the internal carotid artery or the intervertebral foramen of the vertebral artery.A 5 F intermediate catheter was sent to the proximal end of the lesion through the 6 F sheath.After choosing the optimal working angle,a 200 cm microguidewire was used to advance the microcatheter into the distal part of the lesion.A 300 cm microguidewire with the tip shaped into a pigtail was exchanged, and then a conventional balloon was inserted for initial predilation, the diameter of which was 80 % of the diameter of the normal blood vessels,and the length completely covered both ends of the lesion.Under fluoroscopy,the pre-expanded balloon was inflated to the standard pressure and maintained for 60 s,after which the balloon was slowly deflated.The DCB system was delivered to the narrow segment using a microguidewire.The DCB has the same diameter as a conventional balloon and is 2–3 mm longer than a stenotic lesion.The DCB was rapidly inflated to 3 atm, slowly pressurized to the target pressure, and inflated for 1 min.The balloon was then deflated and angiography was performed to rule out dissection,perforation,and distal embolism.After balloon removal,monitoring was continued for 5 min to confirm the absence of complications and the procedure was completed.Possibly persistent residual stenosis was measured by angiography and the antegrade blood flow after inflation was evaluated according to the TICI grading system.Residual stenosis<50%,and TICI grade equal to 3 were defined as successful recanalization.In patients with unsuccessful DCB dilation or blood flow-restricted dissection after drug balloon dilation, bailout stent placement would be performed based on the experience of the operator.Blood pressure was strictly controlled within 72 h after the operation and the blood pressure was maintained at 10%–20%lower than before surgery during the perioperative period.Subjects without stent implantation were administered clopidogrel 75 mg/day and aspirin 100 mg/day for 3 months; subjects with stent implantation were administered clopidogrel 75 mg/day and aspirin 100 mg/day for 6 months.Clopidogrel was subsequently discontinued, and aspirin 100 mg/day was taken for a prolonged period.
2.6.Management of adverse events
All adverse events were treated and followed up during the trial period until the symptoms resolved or stabilized.Adverse events were assessed and documented by the investigator, and the potential relationship between the severity of adverse events and the device was determined.The severity of adverse events or procedural complications were graded, and written statements will be produced by the treatment team.The investigator should report it within 24 h to the relevant ethics committee, Food and Drug Administration Department, and Health and Family Planning Department of the province and autonomous region or municipality directly under the Central Government, where the clinical trial institution is located.Clinical trial institutions and sponsors must also be notified.In the event of death, clinical trial institutions and researchers should provide all necessary information to the ethics committee and sponsors.The investigator should record any device defects found during the course of the clinical trial;analyze the cause of the event together with the sponsor;form a written analysis;propose opinions on the continuation,suspension,or termination of the trial;and report to the clinical trial institution and ethics committee.
2.7.Follow-up and assessment
Assessments were performed by experienced neurosurgeons or interventional physicians and included National Institutes of Health Stroke Scale (NIHSS) and modified Rankin Scale (mRS) (i.e., NIHSS scores for neurological impairment and mRS scores for overall disability).Patients underwent neurological NIHSS assessment(s) and other laboratory examinations on postoperative day 7 or discharge, and were followed up at 30 days,6 months,and 1 year for mRS assessments.Followup imaging was performed to observe whether restenosis occurred in the target vessels.Digital subtraction angiography (DSA) was performed 6 months postoperatively and CTA was performed 1 year postoperatively.Restenosis was defined as>50%stenosis within or immediately adjacent(within 5 mm) to the treated segment and >20 % absolute luminal losscompared to immediate postoperative stenosis.Medication and adverse event records were collected at corresponding time points.Table 3 lists the detailed follow-up and assessment plan.
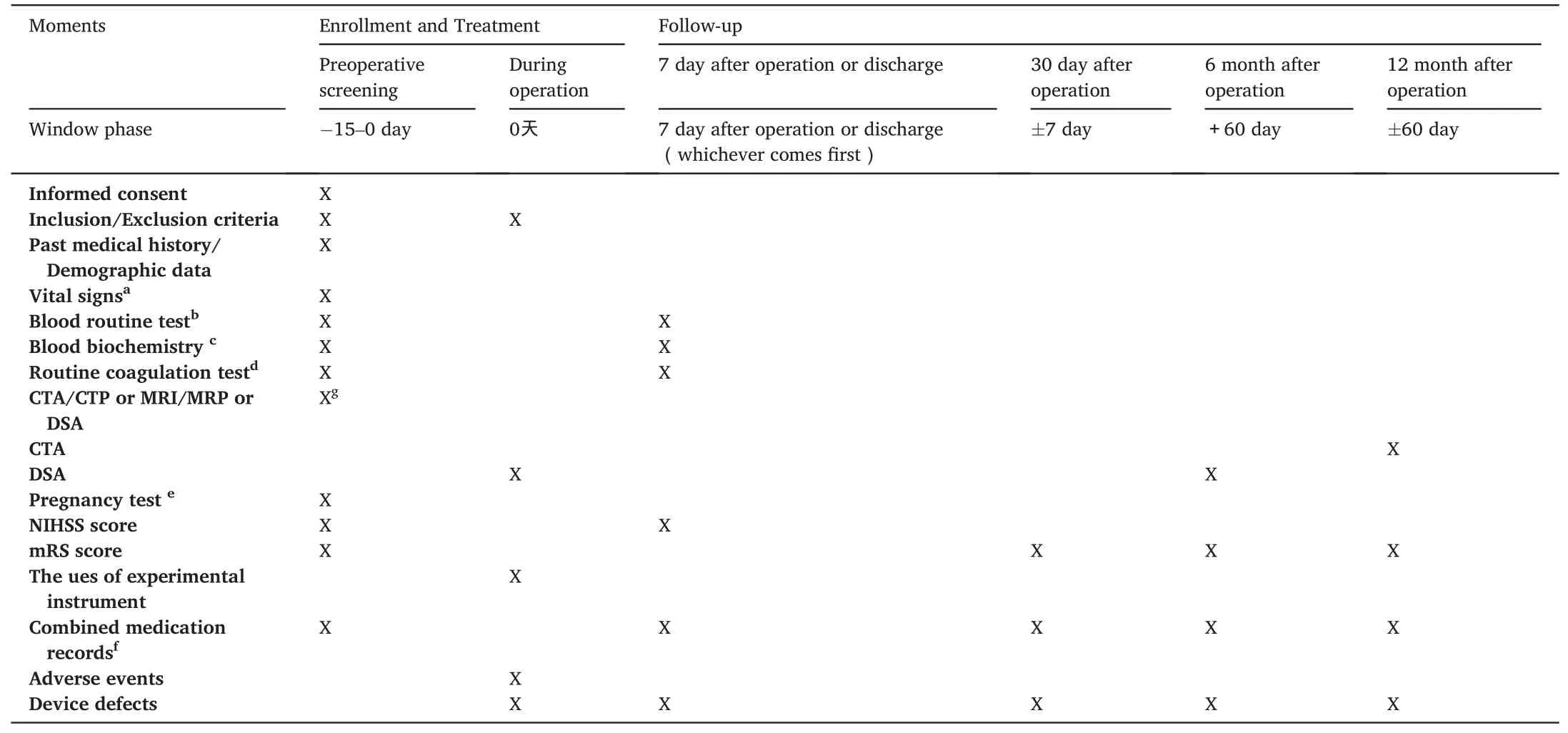
Table 3 Flowchart of the study.
2.8.Data management
All clinical data required for this study, including relevant medical records,imaging data,such as DSA,and other documents,were collected and managed using the Electronic Data Capture(EDC)system developed by the National Cardiovascular Center.Before the system went online,relevant users were required to be trained and tested to ensure that the system met the trial requirements.Subsequently,the individual involved in the trial was assigned an account number and password bound to the user's role and permissions.The researchers ensured the authenticity and integrity of the data and used the EDC to transmit the data to a server over the Internet.
2.9.Reduction and avoidance of bias
In order to prevent the bias of trial results caused by the systematic error of a single center and to reduce the operational differences between centers, we combined case samples from multiple centers to make this experiment more representative and cooperated with the administrators of each trial center to train the investigators on the trial protocol before the start of the clinical trial, so that the investigators could understand and be familiar with the trial products.The experimental personnel at each center were trained on the effectiveness and safety indicators using the assessment criteria.Restenosis rate of the target lesion, the primary endpoint,was evaluated by a third-party independent core laboratory to ensure independent and impartial analysis of the images.Drawing up a supervisory plan, the sponsor appointed qualified monitors to conduct regular on-site monitoring of the trial hospital to ensure that all contents of the research protocol strictly complied with and monitored the case report form(CRF)to ensure that it was consistent with the original data(this study used an electronic case report form).
2.10.Monitoring plan
A data-monitoring committee (DMC) composed of neurointerventionists, neurointensivists, neurologists, and biostatisticians evaluated the accumulated safety data.They were independent of the study sponsor and responsible for monitoring the safety and ethical conduct of the clinical trial.The DMC reviewed and adjudicated all potentially unidentifiable TIA, stroke, and death outcomes.In addition,the DMC was responsible for reviewing clinical event summaries and related imaging studies.All serious adverse events were evaluated by the DMC for their potential relationship to the procedure or study equipment.
2.11.Sample size
A total of 155 patients were enrolled in the present study, and the sample size was calculated based on the restenosis rate of the target lesions 6 months after the operation.According to literature reports and clinical experience,the restenosis rate of target lesions was assumed to be 10%6 months after the trial,and the evaluation standard or target value was set at 20%.When the significance level of the statistical test was set at 0.025 on one side and statistical power was set at 80%, a maximum possible dropout rate of 30%was considered.The sample size calculated according to statistical principles,therefore,was 155 patients.
2.12.Primary outcome
The primary outcome of this study was the target lesion restenosis rate at 6 months (+60 days) postoperatively.Restenosis was defined as>50% stenosis within or immediately adjacent (within 5 mm) to the treated segment and>20%absolute luminal loss compared to immediate postoperative stenosis.The method of estimating the degree of stenosis refers to the method of calculating the stenosis rate in the Warfarin-Aspirin Symptomatic Intracranial Disease(WASID) trial,as follows:
(1– [diameter of the artery at the narrowest part of the stenosis/diameter of the normal artery at the proximal end of the stenosis]) ×100%.
2.13.Secondary outcomes
Secondary outcomes of the trial were as follows: operative success rate judged by immediate postoperative angiography (time frame,immediately after the procedure);success rate of the device;success rate of the procedure;target lesion stenosis(time frame,6 months[±60 days]and 12 months[±60 days]);target lesion restenosis at 12 months[±60 days] after surgery; stroke events (time frame, 30 days [±7 days], 6 months[±60 days]and 12 months[±60 days];ischemic stroke and TIA in the area of the responsible blood vessels (time frame, 30 days [±7 days],6 months[±60 days]and 12 months[±60 days]);ischemic stroke and TIA outside of the area of the responsible blood vessels(time frame,30 days[±7 days],6 months[±60 days],12 months[±60 days]);nonstroke bleeding (time frame, 12 months [±60 days]); mortality (time frame, 30 days [±7 days], 6 months [±60 days] and 12 months [±60 days]);and incidence of serious adverse events and adverse events(time frame,12 months[±60 days]).
2.14.Statistical analysis
Enumeration data will be expressed as frequency and composition ratio, and measurement data will be expressed as mean ± standard deviation,median(25th and 75th percentile),and maximum and minimum values.The baseline demographic analysis will be based mainly on descriptive analysis methods.For the main efficacy indicators, the asymptotic normal method or exact probability method will be used to estimate the restenosis rate and corresponding 95% confidence interval of the target lesion at 6 months after the operation.The upper limit of the confidence interval will be compared with the preset target value to judge the outcome of this trial and whether the product meets the needs of clinical application.The analysis of other efficacy indicators is the same as the baseline.For intra-group comparison of secondary efficacy indicators, the paired t-test will be used for normally distributed measurement data, the Wilcoxon signed-rank test will be used for nonnormally distributed measurement data, and McNemar's paired chisquared test will be used for intra-group comparison of qualitative indicators.Adverse events will be described as the number of cases and incidence rate of adverse events.At the same time, the specific manifestations, degree, and relationship with device implantation of all adverse events will be described in detail.For the primary outcome indicators,statistical analysis will be performed at a one-sided significance level of 0.025 (corresponding to the one-sided confidence limit of the 95%confidence interval),and statistical analysis of other indicators will be performed at a two-sided significance level of 0.05(unless otherwise specified).Statistical analysis will be performed using SAS version 9.4(SAS Institute,Cary,NC, USA).
2.15.Ethics approval and informed consent
This clinical practice complies with the Declaration of Helsinki and the relevant national laws and regulations.This study was approved by the Ethics Committee of Henan Provincial People's Hospital (reference number: 2020-145-03) and other research centers and has been registered in the Chinese Clinical Trial Registry (registration number:ChiCTR2100047223).Participants provided informed written consent.For subjects with impaired consciousness or language comprehension,patients could also enter the trial if the ethics committee agreed in principle and the investigator believed that it was in the patient's interest to participate in the trial.However,informed consent was obtained from their legal guardians before the trial.The subjects had the right to withdraw from the clinical trial at any stage.
3.Outcomes
A total of 155 patients were enrolled in this study.The preliminary collection of follow-up data has been completed, while data quality control is ongoing.
4.Discussion
4.1.Advantages of the Ziyan balloon
To the best of our knowledge,the balloon used in the present study is the first DCB designed specifically for intracranial use and has been clinically validated.Sequent Please NEO (B.Braun, Berlin, Germany) is an early example of a cardiovascular DCB applied to the intracranial arteries.It has a working length of up to 145 cm, which meets the treatment needs of intracranial arterial endpoints.However, it is somewhat rigid and unsuitable for intracranial artery size.14The Ziyan DCB catheter can reach up to 150 cm, and has been made to improve its flexibility.As a specialized balloon for intracranial use, the Ziyan DCB,which has different models in length and diameter, offers a more comprehensive product than the Sequent Please NEO.Moreover, the coating material of the Ziyan DCB is paclitaxel and iohexol,which avoids the introduction of auxiliary materials that may affect drug stability or pose potential biocompatibility risks.
4.2.Why was a single-arm study design used in this study?
The purpose of this trial was to evaluate the safety and efficacy of a new Ziyan DCB catheter for symptomatic, severe ICAS using a prospective, multi-center, single-arm target value approach.The rationale for using a single-arm trial design was that there is currently a lack of industry evaluation standards for the safety and effectiveness of intracranial DCB; however, intracranial DCB therapy is a relatively mature treatment method.15,16In addition, the inclusion standard of diseased arteries in this study was set at ≥l mm,and there are no other proven safe and effective products to serve as controls;as such,we decided to use the single-arm, target-value method for evaluation.Based on previous literature reports addressing intracranial drug balloons and clinical experience, we estimated the restenosis rate within 6 months and ultimately decided to include 155 subjects.
4.3.What are the differences in the inclusion criteria of this clinical trial?
There is no unified standard for the timing of surgery in patients with severely symptomatic ICAS.A multi-center study optimized the inclusion criteria based on the Stenting and Aggressive Medical Management for Preventing Recurrent Stroke in Intracranial Stenosis (SAMMPRIS) trial.Considering plaques of the responsible vessels in the subacute stage,intraoperative procedures are prone to plaque fragmentation and lead to distal embolism and other complications.The criteria for patients to be included 3 weeks after the occurrence of acute stroke were proposed,and this previous trial achieved a good prognostic outcome.17Subsequently,several Chinese studies,including the China Angioplasty and Stenting for Symptomatic Intracranial Severe Stenosis(CASSISS)trial,considered this as an inclusion criterion.However, in the Wingspan Stent System Post Market Surveillance Study (WEAVE), the timing of surgery was 8 days after the last acute ischemic stroke,and adverse events were found to be very low.18The inclusion criteria for this study were that the time interval between the latest ischemic stroke and intravascular intervention should be > 2 weeks because, first, it is based on Chinese expert consensus19that it is safe to perform endovascular treatment with an interval of>2 weeks after the last ischemic stroke.Second,it is based on the experience of our center, which is a better choice for ensuring surgical safety and minimizing economic pressure on patients.In addition,different from some previous inclusion criteria for the diameter of the diseased blood vessel (diameter of 2–4 mm), the Ziyan DCB was considered to be a special intracranial drug balloon, and the range of diameters of intracranial blood vessels was wider than that of coronary arteries20,21;as such,the diameter of the diseased artery in the inclusion criteria was set as ≥l mm.
4.4.Limitations
The present study also had some limitations.For example,due to the one-arm, target-value study design, a randomized control group could not be used to balance out the influence of possible confounding factors.In addition,the main purpose of this study was to explore the short-term efficacy of the Ziyan DCB for the treatment of ICAS;therefore,the followup time was short,but we will continue regular follow-ups after the trial to further study the long-term efficacy of the Ziyan DCB.
5.Conclusion
In conclusion, the purpose of this study was to determine the safety and reliability of a new intracranial DCB.The experimental results not only strongly suggest that this product is effective, but also provide a more solid theoretical basis for the use of drug balloons in treating symptomatic,severe cases of ICAS.
Funding
This trial was funded by the Henan Province Key Research and Development&Promotion Special(Science and Technology)Foundation(Grant/Award Number:222102310378 and 232102231042),the Henan Medical Science and Technology Research Plan Provincial and Ministerial Youth Project(Grant/Award Number: SBGJ 202303001), and the National Health Commission Capacity Building and Continuing Education Project(Grant/Award Number:GWJJ2023100101).
Patient consent
Written informed consent was obtained from patients for publication of these case reports and any accompanying images.
Declaration of competing interest
Yingkun He is the youth editorial board member and Tianxiao Li is the editor-in-chief for Journal of Interventional Medicine,they were not involved in the editorial review or decision to publish this article.All authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
杂志排行

Journal of Interventional Medicine的其它文章
- Mechanisms and therapeutic strategies to combat the recurrence and progression of hepatocellular carcinoma after thermal ablation
- Overview of peripheral arteriovenous malformations: From diagnosis to treatment methods
- Embolization of brain arteriovenous malformations with squid co-polymer embolic material: Initial experience
- Combination of transarterial radioembolization with atezolizumab and bevacizumab for intermediate and advanced staged hepatocellular carcinoma: A preliminary report of safety and feasibility
- Argon-helium cryoablation treatment of undifferentiated pleomorphic sarcoma of the thyroid: A case report and literature review
- “Guidezilla” extension catheter combined with balloon technique for treating pulmonary artery stenosis caused by Takayasu arteritis