矮小并胰岛素抵抗1例及文献复习
2023-03-02卫海燕
刘 英 陈 琼 刘 芳 卫海燕
郑州大学附属儿童医院内分泌遗传代谢科 (河南 郑州 450000)
1975年Gorlin[1]首次报道了一种罕见的常染色体显性遗传病,并依据该疾病临床表现的英文首字母将其命名为SHORT综合征,其典型的临床表现为矮身材(Short stature)、关节过伸和/或腹股沟疝(Hyperextensibility of joints or inguinal hernia)、眼球深陷(Ocular depression)、Rieger畸形(Rieger anomaly)及出牙延迟(Teething delay)。SHORT综合征主要由位于人类第5号染色体的PIK3R1基因突变引起[2–4],目前已报道50余例,国内已报道7例[5–10]。本文报道一例延迟诊治的矮小并胰岛素抵抗,基因结果回示为PIK3R1基因突变所致。
1 病例介绍
患儿,男,14岁,因“生长发育迟缓14年,发现血糖升高9天”入院。患儿14年前(生后)出现生长发育迟缓,出生时身长49cm,体重2.5kg,现14岁7月,身高149.3cm,体重36.5kg(生长曲线见图1)。自幼进食量欠佳,约为同龄儿1/3-1/2,伴出牙延迟、牙列不起,2岁出牙,7岁换牙,平素无头痛、头晕,无腹痛、腹泻,无多饮、多尿等不适,无视觉、听觉障碍。9年前至当地医院行生长激素激发试验,诊断“特发性矮小症”予生长激素(2.0u,qd)治疗2月,身高增长约2.3cm,因出现皮下结节停用,后间断予口服中药治疗,效差。7年前因“自幼身材矮小”至外省某医院就诊,再次行生长激素激发试验,按“特发性矮小症”予注射生长激素治疗9月,身高增长约5.1cm,因监测空腹胰岛素升高停用。因合并特殊面容,曾被疑诊Silver-Russel综合征,行Silver-Russel基因检测及矮小症NGS panel检测,结果提示为阴性,建议定期观察随诊。2年余前再次因“生长发育迟缓”至当地医院住院诊治,诊断“生长发育迟缓查因、胰岛素抵抗”予口服二甲双胍治疗,出院后未规律诊治,未监测胰岛素及血糖情况,仅自行监测生长速率(近2年半身高增长约19cm)。9天前再次至当地医院就诊,查空腹血糖及空腹胰岛素升高,遂来诊。既往史:13年前(1岁时)行腹股沟疝修补术。无传染病接触史,无过敏史、输血史,正常行预防接种。个人史:患者系第1胎第1产,孕40周剖宫产,出生体重2.5kg(-2SD),身长49cm,出生时无抢救史及窒息史。生后母乳喂养,6月添加辅食,1岁断奶,现普通饮食,智力、运动、语言发育与同龄儿相仿。家族史:父母非近亲婚配,父亲身高170cm,体重84kg,母亲身高149cm,体重45kg,PTH:166+5cm。母孕期体健。有1弟弟,8岁,体健。无糖尿病家族史。入院体格检查:身高149.3cm(<-2SD),体重36.5kg(<-2SD),BMI16.37kg/m2(10th),身材矮小,消瘦体型,头发稀疏卷曲,面部脂肪少,三角脸,眼凹陷,鼻翼薄伴鼻小柱,人中短,大耳,唇薄,嘴角下垂,牙列不齐,下颌尖且突出,颈部及腋窝可见黑棘皮症,全身皮下脂肪少,腹部皮下脂肪约0.6cm,心肺腹及神经查体未见异常。双肘关节轻度外翻,双手第五指短,双足第一二趾间距宽。双侧乳房Tanner I期,外生殖器呈男性外观,Tanner III期,阴茎约5cm*2cm,双侧睾丸位于阴囊内,约6~8mL。
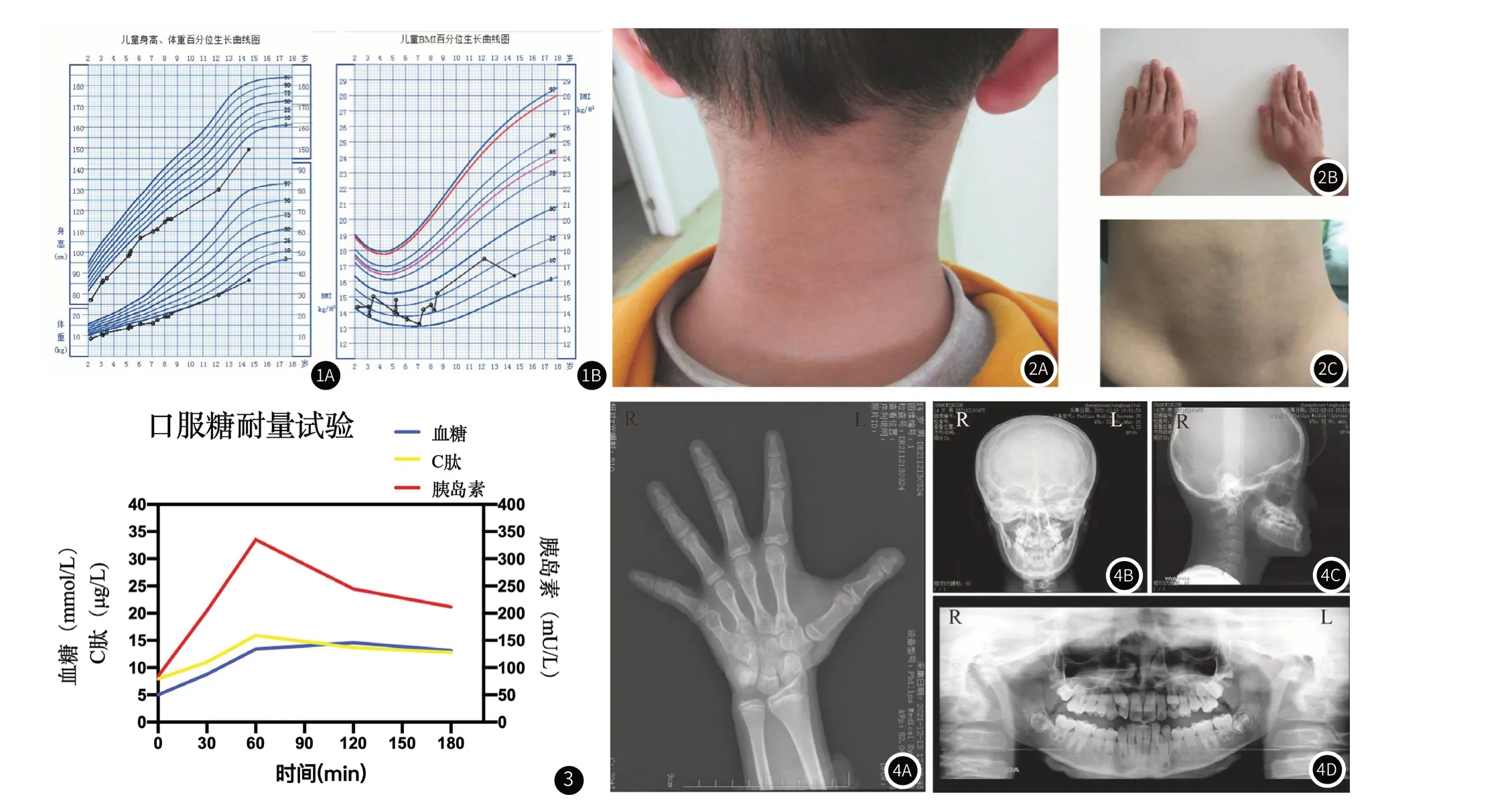
图1 患儿体格生长发育与营养评价。图1A:患儿身高、体重百分位生长曲线图;图1B:患儿BMI百分位生长曲线图。图2 SHORT综合征患儿临床特征 图2A:颈部背侧黑棘皮征;图2B:双手第5指短小;图2C:背侧髋部皮下脂肪菲薄。图3 SHORT综合征患儿口服糖耐量试验。图4 SHORT综合征患儿影像学资料 图4A:患儿左手正位片:掌指骨均短粗,第五掌骨短粗明显;图4B、图4C:头颅正、侧位片;图4D:全口曲面断层片:牙列不齐,乳牙滞留,错合畸形。
实验室检查:矮小症NGS panel检测、Silver-Russell基因检测:阴性。医学外显子组测序基因检测结果:阴性。染色体核型分析:46,XY;葡萄糖:14.7mmol/L(3.9~6.1mmol/L);空腹胰岛素:411μU/mL(2.5~7.1μU/mL)。入院后:血气分析、血氨、血乳酸、血常规、粪常规、肝肾功能、心肌酶、电解质、血脂、免疫功能、肾早损、无机元素、甲胎蛋白、癌胚抗原、促肾上腺皮质激素、皮质醇、甲状腺功能及相关抗体、血尿遗传代谢筛查、心电图、肝脾肾睾丸彩超、胸片、双下肢诸骨均未见异常。尿常规示尿葡萄糖4+>=56mmol/L; HbA1c8.58%(4.8-6.5%);白蛋白46.7g/L,果糖胺(糖化白蛋白)392.77umol/l,GA比值19.58%(10.8~17.1%);谷氨酸脱羧酶自身抗体(GAD)、胰岛素自身抗体(IAA)均阴性;胰岛素样生长因子(IGF-1):442.346ng/mL(226~903ng/mL);OGTT试验示胰岛素抵抗(图3)。左手腕骨片提示骨龄为15岁1月,左手掌指骨均显示短粗,第五掌骨短粗明显。听力检测正常。眼科检查:双侧视力、眼压正常,眼底未见异常。口腔科检查:牙列不齐,乳牙滞留,错合畸形。该患儿影像资料见图4。基因检测:获得家属及患儿知情同意后,抽取患儿及其父母静脉血2mL行基因检测。全外显子组测序结果回示(图5):患儿PIK3R1基因存在c.1945C>T杂合突变,致使第649位氨基酸由精氨酸变为色氨酸(p.R649W),参照美国医学遗传学和基因组学学会(american college of medical genetics and genomics,ACMG)相关指南,该变异为致病变异。经Sanger测序家系验证显示该变异真实可靠,其父母均未携带此突变,推测为新发变异或者父母一方存在生殖细胞嵌合。

图5 SHORT综合征患儿基因检测结果 图5A:患儿基因变异位点;图5B:患儿变异位点验证。
治疗:住院期间予糖尿病饮食、监测血糖、口服二甲双胍等综合治疗,治疗前空腹血糖波动于6.1~15.3mmol/l,餐后血糖波动于11.3~16.4mmol/l;治疗后空腹血糖波动于4.8~7.5mmol/l,餐后血糖波动于5.7~9.2mmol/l;院外随访血糖控制可,糖化血红蛋白6.3%,血糖平均值为7.4mmol/l。
2 讨 论
SHORT综合征(OMIM 269880)是一种罕见的常染色体显性遗传性疾病,其患病率和死亡率尚不清楚。SHORT综合征具有特征性的身材矮小、关节过度伸展和(或)腹股沟疝、眼凹陷、Reiger异常及萌牙延迟,Dyment[2]报道显示仅约40%的患者表现Reiger异常,提示Reiger异常对于诊断SHORT综合征并非必要,另一项回顾性分析[4]显示其4个或4个以上特征性的临床表现仅出现在约52%的患者中。重新评估后显示宫内生长受限(Intrauterine Growth Retardation,IUGR)、皮下脂肪萎缩、胰岛素抵抗和特殊面容亦是SHORT综合征的主要临床特征[2,11]。Mubeen[12]和Masunaga[13]有关SHORT综合征的回顾性研究显示该疾病均有萌牙延迟、眼球凹陷、三角脸和卵巢囊肿表现,约64%的患者出现胰岛素抵抗和糖尿病表现。SHORT综合征其他非特异性临床特征还表现为语言发育延迟、感音神经性耳聋、先天性心脏病等,但智力发育多在正常范围内,学习成绩大都正常[4,14]。此外2020年首次报道了一列合并甲状腺疾病的SHORT综合征,但甲状腺疾病是否为SHORT综合征新的临床表现尚不明确[10]。目前SHORT综合征尚无公认的临床诊断标准,确诊需依赖于基因分析。本例患儿自幼身材矮小,有关节过度伸展和腹股沟疝、眼凹陷、萌牙延迟,存在IUGR、皮下脂肪萎缩、特殊面容,基因检测证实为PIK3R1基因热点突变导致的SHORT综合征。
2013年Dyment[2]研究证实PIK3R1基因突变是SHORT综合征的唯一致病基因,该基因长度为86006bp,位于5q13.1区,包含15个内含子及16个外显子,共编码724个氨基酸。PIK3R1基因通过编码磷脂酰肌醇-3激酶(Phosphatidylin-ositol-3-kinase,PI3K)的调节亚单位(p85α、p55α和p50α),激活蛋白激酶B(protein kinase B,AKT) /雷帕霉素靶体蛋白(mammalian target of rapamycin,mTOR)信号通路,参与胰岛素受体(Insulin Receptor,INSR)、生长激素受体(Growth hormone receptor,GHR)、胰岛素样生长因子1受体(Insulin-like growth factor-1 receptor,IGF-1R)及免疫相关的信号通路传导,从而调节胰岛素信号传导、细胞生长和增殖、脂肪分化等。PIK3R1基因突变导致调节亚基p85α功能缺失,不能与催化亚基p110结合,致使PI3K-AKT-mTOR信号转导通路受损,这一机制可以解释SHORT综合征中IUGR、身材矮小、脂肪营养不良、胰岛素抵抗及糖尿病等临床表现。PIK3R1蛋白的Src-Homology(SH2)结构域在PI3K的调节活性中起重要作用[15]。根据人类基因突变数据库(HGMD)数据,已发现12个可引起SHORT综合征的PIK3R1基因突变,目前已报道的致病变异多聚集在PIK3R1的SH2结构域,其中c.1945C>T突变为SHORT综合征的热点突变,常有典型的临床特征[16]。本例患儿基因检测回示PIK3R1基因存在c.1945C>T这一热点突变,具有胰岛素抵抗、糖尿病及脂肪营养不良表现,但未有Reiger异常的表现,需进一步随访。
研究表明PIK3R1基因突变除引起SHORT综合征外,还可引起2型PI3Kδ过度活化综合征(activated phosphoinositide 3-kinase δ syndrome 2,APDS2)[17],该病为一种罕见的原发性免疫缺陷疾病。Bravo[18]一篇报道显示单一PIK3R1基因突变的患儿同时出现了APDS2和SHORT综合征,因此在诊断为SHORT综合征时需要进一步判断是否存在APDS2。APDS2的临床特点主要为反复呼吸道感染,淋巴组织增生等,免疫功能提示IgM升高,本患儿无反复感染史,免疫功能亦未见异常,因此可排除APDS2。SHORT综合征虽具有特殊面容,但仍需与其他特殊面容相关的综合征进行鉴别,如Silver-Russel综合征(Silver Russell Syndrome,SRS)。SRS[19]是一种罕见的遗传异质性疾病,主要由7号染色体母源性单亲二倍体[UPD7(mat)]和11p15区域母源性或父源性印记基因IGF2和H19表达缺陷所致,临床表现为IUGR、小于胎龄儿、生长发育迟缓、喂养困难、身体不对称以及特殊面容。本例患儿有IUGR及生长发育迟缓,存在特殊面容,就诊过程中曾考虑为该病可能,但该患儿肢体对称,SRS相关基因结果为阴性,基因回示后明确诊断为SHORT综合征。
SHORT综合征可累及多个系统,需要多学科综合管理,在治疗上需要根据个体临床症状及监测情况决定。在婴儿期,因有潜在的心脏畸形(尤其是肺动脉狭窄)及感音神经性听力损失需进行心脏、听力评估;鉴于Rieger异常和青光眼可能的风险,亦需进行眼科评估。在童年时期,应重点进行身高及发育里程碑(尤其是语言发育)的评估。此外因胰岛素及糖尿病的发生,建议每年监测HbA1c、空腹血糖、胰岛素水平,在上述指标正常时亦建议进一步行OGTT试验评估胰岛素抵抗情况[4]。SHORT综合征存在IUGR,童年时期多伴有不同程度的身材矮小,目前生长激素治疗在SHORT综合征中报道较少。生长激素治疗是增加终身高的有效方法,然而生长激素会降低胰岛素敏感性,导致胰岛素抵抗风险增加并加重原有的胰岛素抵抗,加速糖尿病进展,因此对于SHORT综合征患者应谨慎评估生长激素治疗的益处和风险。Thauvin[11]的研究显示SHORT综合征成人终身高在男性为153.7~167cm,女性为 141~160cm。Verge[20]报道一例应用生长激素治疗的SHORT综合征患儿,在其应用生长激素治疗期间,生长速率明显改善,但在应用25个月之后被诊断为糖尿病。本例患者存在IUGR,自幼身材矮小,间断应用生长激素治疗,治疗期间生长速率分别为2.3cm/2月、5.1cm/9月,但先后因出现“皮下结节”及胰岛素抵抗停用,因此生长激素在SHORT综合征患儿中的应用仍需进一步研究。SHORT综合征患儿因存在胰岛素抵抗,胰岛素治疗时用量均偏大[>1.5U/(kg.d)],且在较大剂量时血糖仍难以达标[20]。Zhang[8]报道2例有患有糖尿病的SHORT综合征,初期应用较大剂量胰岛素血糖未控制,调整为二甲双胍治疗后血糖明显改善,提示二甲双胍治疗有效。但其中1例监测胰岛素水平较前升高,这与另一项研究中二甲双胍加重SHORT综合征患儿的胰岛素抵抗相似[21],其具体机制尚不明确。因此二甲双胍在SHORT综合征中的应用需进一步研究并进行长期随访。本例患者使用二甲双胍治疗后空腹及餐后血糖控制达标,但胰岛素水平尚需进一步监测。
SHORT综合征的预后与年龄、累及脏器及其功能、治疗效果有关。大多数情况下,SHORT综合征患儿预期寿命不受影响[8]。但作为一种罕见的常染色体显性遗传疾病,下一代患病的概率为50%,因此,这些患者应常规进行遗传咨询。SHORT综合征临床罕见,容易漏诊、误诊,本文报道矮小并胰岛素抵抗1例,该患儿具有典型SHORT综合征的临床表现及特殊面容,长期就诊过程中均未考虑该疾病可能,提示临床医师应提高对SHORT综合征的认识及治疗,减少漏诊和误诊。
