齐墩果酸-环金属铱配合物的点击化学合成及抗肿瘤性能
2023-02-27李世杰钱晓婷黄元镭薛旭玲刘红科
李世杰 钱晓婷 黄元镭 薛旭玲 钱 勇 苏 志 刘红科
(南京师范大学材料与科学学院,南京 210023)
0 引言
癌症由于具有高死亡率、易复发、侵袭和强转移能力,已经成为威胁全球人类健康的主要公共卫生问题之一[1]。在癌症的临床治疗中,以顺铂为代表的金属药物发挥着独特的作用,并在化疗药物发展的舞台上扮演着重要角色[2]。近些年来,与铂类药物相比,非铂类金属配合物具有多样性的结构、多样化的配位类型、丰富的光学性质及异于顺铂的抗肿瘤作用机理等优良特性,引起了科研工作者们的极大兴趣[3‑8]。其中,环金属铱配合物表现出良好的光物理特性,这使其成为医疗试剂和成像剂的最佳候选者[9]。Mao等通过对环金属铱配合物进行N‑杂环卡宾修饰,实现了特定靶向线粒体的光动力治疗[10]。Chao等通过调节环金属铱配合物的相关配体来调节其亲脂性[11]。本课题组设计的环金属铱与大黄酸相结合后实现了克服顺铂耐药性[12],启发我们将金属前体与天然产物相结合,有望实现金属药物的多功能化应用[13]。
天然产物因其具有可修饰的活性位点、丰富的生物活性以及潜在的靶点等优势,逐渐走入人们的视野。Liang课题组将天然存在的生物碱1,2,3,4‑四氢异喹啉(THIQ)进行修饰后与有机金属Au结合引起内质网应激,诱导癌细胞凋亡和自噬[14]。本课题组在天然产物的金属化修饰方面做了一系列研究,并取得了较大的进展。我们发现,将天然产物与金属配合物连接后能够克服天然产物固有的缺陷。将姜黄素与半夹心Os(Ⅱ)芳烃偶联,开辟了Os‑芳烃配合物的光化学疗法[15]。对天然产物阿魏酸进行化学修饰后,通过酰胺反应与环金属钌连接,使其具有良好的荧光性能[16],将紫苏醇引入到芳基钌配合物中,克服了其剂量大、毒性大的缺点[17]。齐墩果酸(oleanolic acid)是广泛存在于天然植物中的一种提取物,表现出较强的抗肿瘤活性,能抑制多种肿瘤细胞的增殖(如慢性粒细胞白血病细胞、人宫颈癌细胞及肝癌细胞株等),通过调控细胞周期的停滞节点以达到杀死肿瘤的效果[18‑21]。然而,由于齐墩果酸水溶性差、药用剂量大及生物相容性差等缺陷,限制了其在临床上的发展。因此,将其与水溶性、生物相容性好的环金属铱结合,将有可能提高齐墩果酸的成药性,同时也有助于提高配合物的生物活性。铜催化的叠氮−炔环加成(CuAAC)反应是诺贝尔化学奖得主K.Barry Sharpless于2001年提出的,其可在细胞内发生,具有反应速率快、副产物对细胞无毒、条件温和、对水与氧等环境不敏感等特性,已广泛应用于生物和化学领域[22]。
在本研究中,我们将齐墩果酸、环金属铱分别通过化学修饰,制备出含有炔基或叠氮基团的前体,通过CuAAC反应得到了新型配合物。通过现代分析手段对化合物组成及脂溶性进行了表征和测试,同时研究了细胞毒性及抗癌机理。
1 实验部分
1.1 实验试剂和仪器
三氯化铱水合物(IrCl3·H2O)、4‑叠氮基甲基‑4′‑甲基‑2,2′‑联吡啶(N3‑bpy)根据文献路线合成[23]。其余试剂均为购买后直接使用,未进行任何提纯。齐墩果酸、二氧化硒、五水合硫酸铜(CuSO4·5H2O)、抗坏血酸钠、2‑苯基吡啶、炔丙胺、1‑(3‑二甲氨基丙基)‑3‑乙基碳二亚胺盐酸盐(EDCI)、1‑羟基苯并三氮唑(HOBT)、硼氢化钠、叠氮化钠购于天津希恩思。其余试剂购于南京晚晴。牛血清白蛋白(BSA)、Dulbecco′s Modified Eagle Medium 细 胞 培 养 基(DMEM)、双抗(链霉素和青霉素)、胰蛋白消化酶、An‑nexin V‑FITC细胞凋亡检测试剂盒、2′,7′‑二氯荧光素二乙酸盐(H2DCF‑DA)试剂盒等生物试剂均购于南京凯基生物。
1H NMR波谱使用Bruker Avance Ⅱ 400 MHz光谱仪检测。电喷雾电离质谱(ESI‑MS)使用LCQ光谱仪(Thermo Scientific)质谱仪检测。UV‑Vis光谱使用Lambda 365紫外可见分光光度计测得。荧光光谱使用FS5荧光分光光度计(Edinburgh Instrument)获得。细胞毒活性通过多功能酶标仪LabServ K3测定。用激光共聚集显微镜(A1,Nikon)测试配合物在细胞内的共定位成像。应用流式细胞仪(BD FACSverse,美国)进行细胞凋亡、周期及活性氧等分析测定。
1.2 配体及配合物的合成
1.2.1 配体OA⁃alkyne的合成
在冰浴和氩气保护条件下,将齐墩果酸(0.91 g,2.0 mmol)与 EDCI(0.38 g,4 mmol)溶于 50 mL 无水DMF中,缓慢滴加300µL三乙胺,并在0℃下搅拌20 min,随后加入含有HOBt(0.21 g,1.6 mmol)的无水DMF溶液,继续搅拌20 min。最后,加入炔丙胺(0.13 g,2.4 mmol),反应16 h。然后加入50 mL水,用二氯甲烷(3×20 mL)萃取,合并有机层,用无水硫酸钠干燥,旋蒸除去二氯甲烷,粗产物经柱层析纯化(DCM/MeOH,200∶1,V/V),得到0.47 g白色固体产物OA⁃alkyne(产率 50%)。1H NMR(400 MHz,DMSO‑d6):δ 7.68(q,J=5.7 Hz,1H),5.21(d,J=3.4 Hz,1H),4.30(d,J=5.1 Hz,1H),3.88~3.70(m,2H),3.03~2.97(m,1H),2.96(t,J=2.5 Hz,1H),2.79(dd,J=13.6,4.5 Hz,1H),1.93~1.20(m,21H),1.14~1.02(m,6H),0.87(dd,J=11.1,7.6 Hz,16H)。
1.2.2 桥联配体N3⁃bpy的合成
在氩气的保护下,向150 mL 1,4‑二氧六环中加入4,4′‑二甲基‑2,2′‑二联吡啶(8.0 g,43.0 mmol)和二氧化硒(8.0 g,71 mmol),将该混合物回流24 h。使其冷却到室温,过滤,旋转蒸发,干燥,加入三氯甲烷,得到黄色固体,将所得的固体溶于50 mL甲醇中,并置于冰浴中,继续用溶解有2.8 g NaBH4的20 mL氢氧化钠(0.1 mol·L−1)溶液滴加到上述混合物中,在室温下搅拌1 h,旋转蒸发除去甲醇,剩余的水溶液用饱和碳酸氢钠溶液稀释后用三氯甲烷萃取。用无水硫酸钠干燥,旋转蒸发除去溶剂,并通过柱层析法纯化(DCM/MeOH,110∶1,V/V),得到白色固体L1(4.0 g,47%)。
在40 mL的48%溴化氢溶液中添加上述获得的L1(2.0 g,10.0 mmol),再将10 mL浓硫酸加至该溶液中,回流过夜。待冷却到室温后,将该混合物倒入100 mL的冰水中,用碳酸钠调整pH值到8.0,然后用三氯甲烷萃取无色的有机层,然后用无水硫酸钠干燥,旋转蒸发除去溶剂;经柱层析纯化(DCM/MeOH,50∶1,V/V),获得 L2(2.4 g,93%)。将 L2(5 g,19 mmol)和叠氮化钠(6.4 g,86 mmol)溶解在 DMF/H2O(10∶1,V/V,50 mL)中,并将混合物在70 ℃下搅拌过夜,真空除去溶剂,用二氯甲烷萃取并用水洗涤,无水硫酸钠干燥后除去溶剂,用柱色谱纯化(DCM/TEA,40∶1,V/V)后得到白色油状固体N3⁃bpy(4.0 g,87%)。1H NMR(400 MHz,DMSO‑d6):δ 8.68(dd,J=4.9,0.8 Hz,1H),8.55(dd,J=5.0,0.8 Hz,1H),8.38(dd,J=1.7,0.9 Hz,1H),8.26(dt,J=1.7,0.9 Hz,1H),7.44~7.41(m,1H),7.31(ddd,J=5.0,1.8,0.8 Hz,1H),4.70(s,2H),2.42(d,J=0.7 Hz,3H)。
1.2.3 CycloIr⁃N3的合成
将 IrCl3·H2O(0.31 g,1 mmol)与2‑苯基吡啶(ppy,0.62 g,4 mmol)溶解在 2‑乙氧基乙醇(20 mL)和水(10 mL)混合溶剂中,加热回流过夜。反应完成后,冷却至室温过滤,再将沉淀物溶于40 mL二氯甲烷中离心,取上清液。将20 mL的甲苯和10 mL的正己烷添加到滤液中,旋蒸将溶剂量减少至10 mL,在4℃冰箱静置一晚,获得[Ir(ppy)2Cl]2。在氩气保护条件下,称取[Ir(ppy)2Cl]2(1.07 g,1.0 mmol)和 N3⁃bpy(0.55 g,2.2 mmol)溶解于二氯甲烷与甲醇(3∶1,V/V,24 mL)中,将混合物在40℃下回流搅拌过夜。旋转蒸发去除溶剂,将固体溶于15 mL甲醇,向上述甲醇溶液中缓慢加入饱和六氟磷酸铵溶液,收集得到的黄色固体沉淀,用硅胶柱层析(DCM/MeOH,100∶1,V/V),得到0.61 g黄色固体产物CycloIr⁃N3,产率约为35%。1H NMR(400 MHz,CDCl3):δ 8.60(d,J=1.7 Hz,1H),8.54(s,1H),7.96~7.88(m,3H),7.82~7.75(m,3H),7.70(dt,J=8.0,1.5 Hz,2H),7.54(ddd,J=7.5,5.8,1.3 Hz,2H),7.43(dd,J=5.7,1.6 Hz,1H),7.23~7.20(m,1H),7.06(dtdd,J=13.1,7.1,5.7,1.4 Hz,4H),6.93(tdd,J=7.5,3.2,1.3 Hz,2H),6.32(ddd,J=7.7,3.4,1.2 Hz,2H),4.81(s,2H),2.62(s,3H)。MS(m/z):[CycloIr⁃N3−PF6]+理论值726.2,实验值726.1。
1.2.4 配合物CycloIr⁃OA的合成
在氩气保护下,将配合物CycloIr⁃N3(0.029 g,0.04 mmol)、配体 OA⁃alkyne(0.024 g,0.05 mmol)、CuSO4·5H2O(0.002 g,0.01 mmol)与 抗 坏 血 酸 钠(0.001 g,0.01 mmol)加入到5 mL无水DMF中,在室温和黑暗条件下,将混合物搅拌8 h,旋蒸除去溶剂DMF,通过硅胶柱层析纯化(DCM/MeOH,200∶1,V/V),得到黄色配合物 CycloIr⁃OA(0.017 g,32%)。1H NMR(400 MHz,DMSO‑d6):δ 8.86(dd,J=19.8,1.7 Hz,1H),8.31~8.22(m,2H),8.00(d,J=9.3 Hz,1H),7.97~7.76(m,6H),7.70(d,J=5.6 Hz,1H),7.66~7.51(m,3H),7.17(dddd,J=14.5,11.5,5.8,1.5 Hz,3H),7.01(tdd,J=7.2,5.8,1.3 Hz,2H),6.95~6.82(m,2H),6.23~6.10(m,2H),5.83(d,J=5.2 Hz,2H),5.13(q,J=3.1 Hz,1H),4.33(d,J=5.0 Hz,1H),4.24(q,J=7.5,5.4 Hz,2H),2.97(t,J=7.0 Hz,1H),2.77~2.71(m,1H),2.55(s,3H)。MS(m/z):[CycloIr⁃OA−PF6]+理论值 1 220.62,实验值1 219.65。
1.3 配合物的光物理性质检测
使用紫外可见分光光度计检测配合物前体和配合物的紫外可见吸收光谱。将配体和配合物溶于 DMSO 中,配制出 20 mmol·L−1的浓储液,再用Milli‑Q超纯水将配合物前体和配合物的浓储液稀释成 30 µmol·L−1的样品溶液(H2O/DMSO,99∶1,V/V),并在实验时将吸收波长采集范围设为250~600 nm。
使用荧光分光光度计检测前体CycloIr⁃N3和配合物CycloIr⁃OA在298 K下的荧光发射光谱。用水将前体CycloIr⁃N3和配合物CycloIr⁃OA的浓储液分别稀释成10 µmol·L−1的样品溶液(H2O/DMSO,99∶1,V/V),并记录下400~800 nm范围内的发射光谱(激发波长为 405 nm)。
1.4 细胞毒性研究
用 MTT 法对 OA⁃alkyne、CycloIr⁃N3、配合物CycloIr⁃OA以及阳性对照药物顺铂(cisplatin)进行细胞毒活性测定。选取人源癌细胞A2780、A549、HeLa以及正常人肺成纤维HLF细胞。细胞在DMEM中培养。将对数生长期的细胞接种在96孔板(每孔5 000个)中,在CO2体积分数5%、310 K条件下培养过夜,然后加入不同浓度的待测药物(DMEM/DMSO,99∶1,V/V)继续孵育48 h。每孔加入20 µL MTT(5 mg·mL−1),继续孵育4 h。小心地将培养液吸出,溶解在150µL的DMSO中,使用LabServ K3酶标仪进行振荡混匀后,读取590 nm处吸光度值,计算半抑制浓度(IC50)。
1.5 配合物的脂溶性测试
使用摇瓶法[24]测试并计算 CycloIr⁃N3和CycloIr⁃OA的脂水分配系数lg Po/w。将等体积的正辛醇与0.9%的氯化钠溶液振摇2 d,使两相之间混合充分,将配体和配合物分别溶解在氯化钠溶液中,然后加入等量的正辛醇,在37 ℃、500 r·min−1下摇动6 h,将样品在 8 000 r·min−1的转速下离心 8 min,用分液漏斗将两相分离,使用紫外可见分光光度计分别测试有机相和水相中的吸光度,利用对数求出lg Po/w。最后结果用3次独立实验的平均值表示。
1.6 配合物的细胞定位检测
分别使用线粒体探针Mito‑green,溶酶体探针Lyso‑blue和细胞核探针Hoechst33342检测A2780细胞的3种亚细胞器中CycloIr⁃OA的分布情况。将A2780细胞以5×105mL−1的密度接种于4孔板中,在CO2体积分数5%、310 K条件下孵育12 h,以保证其贴壁生长。然后向其中加入含2 µmol·L−1CycloIr⁃OA的DMEM培养基(DMEM/DMSO,99∶1,V/V)溶液继续孵育12 h。将培养基吸出,用磷酸盐缓冲溶液(PBS)洗涤 3 次,分别加入 2 µmol·L−1探针避光孵育 30 min,用PBS洗涤3次,在共聚焦显微镜下进行测试。
1.7 配合物诱导细胞凋亡检测
利用细胞凋亡检测试剂盒探究配合物CycloIr⁃N3和CycloIr⁃OA诱导A2780细胞的死亡方式。在6孔板中接种上密度为1.5×106mL−1的A2780细胞,在CO2体积分数5%、310 K条件下孵育12 h,以保证其贴壁生长。然后向每孔中加入只含有1%DMSO的DMEM培养基(DMEM/DMSO,99∶1,V/V)以及含有不同浓度梯度的配合物培养基继续孵育24 h。收集细胞,用1×PBS缓冲液清洗细胞2次,加入500µL的Binding buffer,重悬细胞。将5µL的V‑FITC和5µL的PI加入到每个孔中后,在黑暗条件下染色20 min,并使用流式细胞仪对样品进行检测。
1.8 配体和配合物诱导细胞内活性氧检测
将 A2780 细胞以 1.5×106mL−1的密度接种于 6孔板中,在CO2体积分数5%、310 K条件下孵育12 h。然后向其中加入只含有DMSO的DMEM培养基(DMEM/DMSO,99∶1,V/V)以及含有不同浓度梯度CycloIr⁃N3和CycloIr⁃OA的培养基继续孵育12 h,用1×PBS缓冲液洗涤3次,在避光条件下,加入无血清DMEM稀释的H2DCF‑DA探针(10 µmol·L−1)孵育25 min。收集细胞,用1×PBS洗涤2次,加入500µL并重悬。通过流式细胞仪在对样品进行分析,并使用FlowJo 10.1处理数据。
将A2780细胞以5×105mL−1的密度接种于共聚焦小皿中,在CO2体积分数5%、310 K条件下孵育12 h,接着将含2 µmol·L−1待测药物的 DMEM培养基(DMEM∶DMSO,99∶1,V/V)溶液加入到皿中,继续孵育12 h。在避光条件下,使用H2DCF‑DA(10µmol·L−1)探针染色 25 min,用 1×PBS 缓冲液洗涤 2次,通过共聚焦成像进行监测。λex=488 nm,λem=(510±20)nm。
1.9 配合物阻滞细胞周期检测
使用细胞周期检测试剂盒研究配合物CycloIr⁃N3和CycloIr⁃OA对A2780细胞周期的影响。在6孔板中接种上密度为1.5×106mL−1的A2780细胞,在CO2体积分数5%、310 K条件下孵育12 h。接着将含有不同浓度梯度的配合物孵育细胞24 h。收集细胞,用1×PBS(4℃)洗涤2次,加入体积分数为70%的冰乙醇来重悬细胞,并置于4℃的冰箱中固定过夜。将固定的细胞离心收集细胞(5 000 r·min−1,5 min,4 ℃),并将染色液(PI/RNase A,9∶1,V/V)加入到细胞中继续孵育30 min。用流式细胞仪对样品进行检测(λex=488 nm,λem=620 nm)。
2 结果与讨论
2.1 合成与表征
选取齐墩果酸进行化学修饰得到炔端配体OA⁃alkyne[25],将桥联配体 N3⁃bpy 与铱二聚体[Ir(ppy)2Cl]2进行反应得到含有叠氮端的环金属铱配合物前体CycloIr⁃N3,将CycloIr⁃N3与OA⁃alkyne在铜催化的条件下进行CuAAC反应[26],得到新配合物CycloIr⁃OA,合成路径如图1所示,并通过1H NMR及 ESI‑MS 对配合物进行了表征(图 S1~S4,Support‑ing information)。

图1 通过CuAAC反应合成CycloIr⁃OA的路线Fig.1 Synthetic routes for complex CycloIr⁃OA
2.2 配合物前体和配合物的光学性质
金属配合物所具有的光物理性质有助于实现在体内进行跟踪检测,并进一步探索其在细胞中的生理功能[27]。图 2A、2B 分别为前体 CycloIr⁃N3、配合物CycloIr⁃OA的紫外吸收及荧光发射光谱。CycloIr⁃N3的紫外吸收波长主要在250 nm,荧光发射波长在600 nm,这与CycloIr⁃OA的紫外吸收和荧光发射波长基本吻合。CycloIr⁃OA在250~450 nm的吸收峰归属于金属到配体的电荷转移(1MLCT及3MLCT),及配体到配体的电荷转移(LLCT)[28]。CycloIr⁃OA用405 nm的光激发时,在600 nm处有较强的荧光发射峰,为在体内进行可视追踪提供了可能。

图2 CycloIr⁃N3(A)及CycloIr⁃OA(B)在298 K时水溶液(20 µmol·L−1)中的紫外可见吸收和荧光发射谱图Fig.2 UV‑Vis absorption and emission spectra of 20 µmol·L−1solution of ligand CycloIr⁃N3(A)and complex CycloIr⁃OA(B)at 298 K
2.3 配合物的脂水分配系数
药物的亲水性或亲脂性过高或低会对药效产生不利影响[29]。这种性质常用lg Po/w表示,其值越大,化合物越容易透过细胞膜进入细胞内,反之化合物易溶于水。我们使用摇瓶法对CycloIr⁃N3和CycloIr⁃OA的脂溶性进行了测试,并用lg Po/w说明化合物的亲脂性能。CycloIr⁃N3和 CycloIr⁃OA 的lg Po/w分别为1.82±0.03和0.8±0.05,这表明引入齐墩果酸后有效加强了配合物CycloIr⁃OA的水溶性,克服了齐墩果酸原有的水溶性差等缺陷,使其与人体环境更好地适配,增加了药物的吸收速度。
2.4 细胞毒性活性研究
采用MTT法测定了OA⁃alkyne、CycloIr⁃N3和CycloIr⁃OA对肿瘤细胞A2780、A549、HeLa及正常细胞HLF的细胞毒性,以顺铂为对照化合物。由IC50值(表1)可以看出,配体OA⁃alkyne对上述肿瘤细胞表现出较低毒性,其中对A2780细胞的IC50为40.5 µmol·L−1;CycloIr⁃N3对上述肿瘤细胞均表现出较好的抗肿瘤活性,尤其对A2780细胞的毒性最好,IC50低至 5.6 µmol·L−1。而当两者通过CuAAC 反应结合后,得到的新配合物CycloIr⁃OA对上述肿瘤细胞的毒性有了明显的提升,优于临床药物顺铂:对A2780 细 胞 的 IC50仅 为 1.6 µmol·L−1,比 前 体CycloIr⁃N3的活性提高了2.5倍,仅为顺铂的IC50的五分之一。这一结果表明,天然产物齐墩果酸的引入可以较大地提升金属配合物的抗肿瘤活性,为高活性抗癌金属药物的研制提供了新途径。由于配合物CycloIr⁃OA对A2780细胞的抗癌活性最佳,我们选用A2780细胞进行抗癌作用机理研究。

表1 化合物OA⁃alkyne、CycloIr⁃N3、CycloIr⁃OA对癌细胞A2780、A549、HeLa和正常细胞HLF 48 h的IC50值Table 1 IC50values of OA⁃alkyne,CycloIr⁃N3,and CycloIr⁃OA against A2780,A549,HeLa cancer,and normal HLF cell lines for 48 hµmol·L−1
2.5 配合物的亚细胞器分布
激光共聚焦成像是探索药物亚细胞器定位常用的方法[30]。配合物CycloIr⁃OA具有良好的荧光性质,因此我们使用3种商用的细胞器探针Mito‑green( 线 粒 体)、Lyso‑blue(溶 酶 体) 及Hoechst33342(细胞核),通过共聚焦显微镜分析了CycloIr⁃OA在A2780细胞内的分布情况。如图3所示,CycloIr⁃OA的红色荧光与线粒体探针Mito‑green的荧光信号有较好的重叠,处理分析后得到CycloIr⁃OA与线粒体共定位系数为0.92,与溶酶体的共定位系数为0.52,基本不定位在细胞核,结果说明CycloIr⁃OA靶向A2780细胞的线粒体。线粒体是细胞和生物体内稳态的重要调节者,涉及许多复杂的癌症生物学过程,如细胞信号传递、细胞代谢和细胞死亡等。因此,线粒体成为抗肿瘤的重要靶点[31]。靶向线粒体是一种可行、有效和安全的抗肿瘤靶向策略。

图3 配合物CycloIr⁃OA与A2789细胞于37℃孵育24 h后的共聚焦显微镜图,图中可以看到CycloIr⁃OA主要分布在线粒体Fig.3 Confocal micrograph of complex CycloIr⁃OA and A2789 cells incubated at 37 ℃ for 24 h shows that CycloIr⁃OA is mainly distributed in mitochondria
2.6 配合物CycloIr⁃OA诱导细胞内活性氧的产生
正常细胞通过控制细胞内活性氧(ROS)水平来调控细胞内信号传导、细胞增殖及细胞死亡等生理过程。据文献报道,促进癌细胞产生ROS是金属药物杀伤肿瘤细胞的作用机制之一[32]。因此为了进一步研究配合物CycloIr⁃OA诱导细胞死亡的作用机理,我们使用商业化的ROS探针H2DCF‑DA检测了肿瘤细胞中的ROS水平[33]。细胞中产生的ROS可以将探针氧化为有荧光的DCF,可以通过观察荧光强度表示细胞内ROS水平。CycloIr⁃N3处理后A2780细胞的绿色荧光强度没有明显增强,流式细胞术结果也证实了CycloIr⁃N3处理后细胞内ROS增加并不明显(图S5)。CycloIr⁃OA处理后的A2780细胞与未加药的对照组相比绿色荧光强度增强(图4A),表明齐墩果酸的引入可诱导细胞产生过量的ROS。流式细胞术也证实了CycloIr⁃OA处理后细胞内ROS的增加(图4B),并呈现出配合物的浓度依赖性,说明ROS的产生可能是诱导癌细胞死亡的原因之一。

图4 CycloIr⁃OA诱导A2780细胞产生ROS:(A)CycloIr⁃OA(2 µmol·L−1)处理A2780细胞12 h后ROS生成的共聚焦图像;(B)CycloIr⁃OA(control和1、2、4 µmol·L−1)处理A2780细胞12 h后ROS生成的流式细胞图Fig.4 ROS generation induced by CycloIr⁃OA in A2780 cells:(A)confocal images of ROS generation in A2780 cells treated with CycloIr⁃OA(2 µmol·L−1)for 12 h;(B)ROS generation in A2780 cells treated with different concentrations of CycloIr⁃OA(control and 1,2,4 µmol·L−1)for 12 h by flow cytometry
2.7 配合物诱导A2780细胞周期阻滞和凋亡
通过流式细胞仪研究了CycloIr⁃OA对细胞周期的影响,结果如图5所示。与未加药物的对照组相比,当将不同浓度的CycloIr⁃OA孵育A2780细胞24 h后,配合物诱导的S期的细胞比例随药物浓度的增大而增大;用6 µmol·L−1CycloIr⁃OA处理后S期细胞数量由13.2%增长到34.9%,而G0/G1期细胞数量从64.5%减少至38.4%,说明CycloIr⁃OA以浓度依赖的方式将细胞周期阻滞在S期,从而诱导细胞死亡。经不同浓度前体配合物CycloIr⁃N3处理后的细胞,随着药物浓度的增加,未能对A2780细胞起到阻滞作用(图S6)。上述结果说明将齐墩果酸引入到环金属铱中,增强了其对细胞阻滞的效果。

图5 CycloIr⁃OA(control和2、4、6 µmol·L−1)和A2780孵育24 h后并用PI染色,通过流式细胞术测定细胞周期Fig.5 Cell cycle assay of A2780 cells by flow cytometry after incubation with CycloIr⁃OA(control and 2,4,6 µmol·L−1)for 24 h and stained with PI
细胞凋亡揭示了一种广泛的免疫病的致病机理,尤其是凋亡与肿瘤的相关性越来越受到关注[34‑35]。通过流式细胞仪对配合物CycloIr⁃OA诱导细胞凋亡的情况进行检测,结果如图6所示。在二维散点图上,Annexin V是x轴,PI是y轴,十字门将图片分为4个象限:左上象限Q1区域的细胞为坏死细胞;右上象限Q2区域的细胞为晚期凋亡细胞;右下象限Q3区域的细胞为早期凋亡细胞;左下象限Q4区域的细胞为活细胞[36]。可以看出,随着浓度的增加,处于坏死区域的细胞比例在逐渐增加。在用4 µmol·L−1的药物处理24 h后,坏死细胞的含量由2.20%增加到75.2%。而经同样浓度的CycloIr⁃N3处理后,坏死细胞含量仅由9.34%增加到12.1%(图S7),由此可见,环金属铱连接齐墩果酸后,使肿瘤细胞坏死数量有了明显提高。因此,CycloIr⁃OA主要以诱导肿瘤细胞坏死的方式导致细胞的死亡。
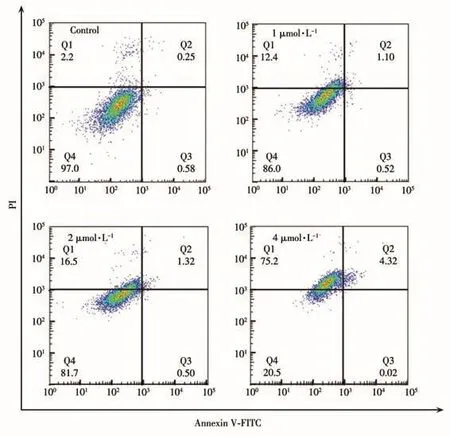
图6 CycloIr⁃OA(control和1、2、4 µmol·L−1)和A2780孵育24 h并用Annexin V‑FITC和PI染色后,通过流式细胞术进行凋亡测定Fig.6 Apoptotic assays of A2780 cells by flow cytometry after incubation with CycloIr⁃OA(control and 1,2,4 µmol·L−1)for 24 h and stained with Annexin V‑FITC and PI
3 结论
我们通过点击反应将无毒性的五环三萜类化合物齐墩果酸引入到环金属铱配合物中,配合物CycloIr⁃OA的抗肿瘤活性有较大提升,明显优于临床药物顺铂,对A2780细胞的IC50为1.6 µmol·L−1,仅为顺铂的五分之一。CycloIr⁃OA主要富集在线粒体中,通过产生大量活性氧杀伤肿瘤细胞,并将肿瘤细胞周期阻滞在S期,诱导肿瘤细胞的坏死。将天然产物引入到金属前体中,不仅提高了配合物的抗肿瘤活性,而且拓展了天然产物在金属药物中的应用,为新型高活性金属配合物的设计、合成提供了指导。
Supporting information is available at http://www.wjhxxb.cn
