高通量计算二维材料界面摩擦*
2023-02-18崔子纯杨莫涵阮晓鹏范晓丽周峰3刘维民3
崔子纯 杨莫涵 阮晓鹏 范晓丽† 周峰3) 刘维民3)
1) (西北工业大学材料学院,先进润滑与密封材料中心,西安 710072)
2) (西北工业大学,伦敦玛丽女王大学工程学院,西安 710072)
3) (中国科学院兰州化学物理研究所,固体润滑国家重点实验室,兰州 730000)
建立了基于第一性原理方法研究二维材料界面摩擦的高通量计算程序,该程序实现了自动化批量建模、批量提交任务、多任务并发计算,以及计算结果自动收集、处理和图像绘制,使用该程序可以节省时间.采用此程序计算了不同层间距离下双层氮化硼和双层石墨烯的滑移势能面,及层间界面摩擦力和摩擦系数.研究发现,随着层间距离减小,双层氮化硼界面的平均摩擦力近似线性增大,摩擦系数为0.11—0.17,双层石墨烯界面摩擦力先增大后减小再增大,其摩擦系数在12 nN 载荷下达到最小值(0.014),这些结果与已有研究结果一致,验证了该计算程序的可靠性.此外还研究了表面氢化和氟化对双层氮化硼界面摩擦的影响,发现氟化氮化硼/氮化硼界面的摩擦系数更低.
1 引言
摩擦一般发生在相对运动或具有相对运动趋势的接触界面,阻碍相对运动,同时产生能量损耗.Holmberg 等[1]调查发现卡车等重型交通工具有1/3 的燃料用于克服发动机、变速器的摩擦.为了避免不必要的能源消耗和机器故障,控制摩擦力、黏附力和磨损力以减小摩擦磨损一直是科研工作者密切关注的问题[2].纳米尺度下,以黏附和摩擦为代表的表面力成为制约纳米机电系统性能和寿命的关键因素[3],因此,了解纳米级甚至原子尺度下的摩擦行为必要且重要.
因为层间作用力弱、层内共价键强、剪切应力较低的特点,层状二维材料的润滑性能往往优于其他纳米材料[4,5].特别是石墨烯[6]、氮化硼[7]、二硫化钼[8]等典型二维层状材料,通常以微米级薄膜的形式被用作固体润滑剂.已有研究表明,二维材料界面处的原子尺度摩擦行为受到多种因素影响,包括温度[9−11]、滑动速度[12]以及载荷[12−14]等.研究者采用实验技术和理论计算方法研究了二维材料界面的原子尺度摩擦行为[15−17].温度变化会影响表面原子热振动的幅度以及频率,从而影响摩擦行为,已有研究显示摩擦系数随温度变化呈现非单调变化的特点[18].摩擦力与滑动速度的关系较为复杂,当滑动速度较小时,摩擦力几乎不随速度变化,能量耗散主要来自于黏滑运动,当滑动速度增大到一定程度时,摩擦力与速度呈现线性关系,黏滑运动消失,能耗主要为阻尼能耗[18−20].
依照宏观摩擦定律,摩擦力与载荷的关系符合阿蒙顿定律,原子尺度上摩擦力与载荷的关系则较为复杂.Mate 等[21]利用钨探针研究了石墨表面的微观黏滑现象,发现界面间的平均摩擦力随法向载荷的增大而近似线性增大;Mo 等[22]采用分子动力学方法模拟了碳尖端在金刚石表面的滑动行为,当接触界面的范德瓦耳斯吸附力较强时,摩擦力和载荷呈现非线性关系,随着范德瓦耳斯吸附力的减弱,摩擦力与载荷的关系趋于线性.An 等[23]基于第一性原理方法计算了氮化硼层间摩擦,发现摩擦力随载荷的增大而增大;Sun 等[24]发现石墨烯层间平均滑移摩擦力随着载荷增大,先增大后减小再增大的有趣现象.此外,多项研究表明[25−30],层数、表面吸附、面内应变等对石墨烯和氮化硼的层间摩擦有着显著影响,归因于层间电子分布的变化.鉴于原子尺度摩擦学的复杂性和重要性,从原子水平上掌握摩擦机制[31],预测和控制摩擦行为紧迫且关键.
原子力显微镜(atomic force microscope,AFM)研究扫描探针与材料表面之间的摩擦特性,是研究纳米尺度摩擦学应用最广泛的工具.然而,受限于可用作探针尖端的材料种类,AFM 测量任意二维材料界面间摩擦仍然是一个挑战.先进的计算模拟技术是研究二维材料层间摩擦的有力工具.研究摩擦现象的常用计算模拟方法包括分子动力学模拟(molecular dynamics,MD)方法和基于密度泛函理论(density functional theory,DFT)的第一性原理方法.分子动力学依据牛顿力学定律模拟接触界面的相对运动,根据公式(U代表原子间相互作用势,r代表原子的所在位置)求出每个原子的受力,然后分别对所有原子的法向力和与滑动方向相反的横向力进行求和,得到总的法向力和摩擦力,进而运用阿蒙顿定律求得摩擦系数.分子动力学模拟已被广泛应用于计算二维材料摩擦学性能[32−35],其模拟的可靠性强烈依赖于选取的势函数描述层间/内相互作用的准确性.第一性原理计算方法通过计算摩擦界面相对滑动时的能量变化,得到滑动能垒和平均摩擦力,通过改变层间距离模拟外界施加载荷,获得不同载荷下的势能面以及摩擦力,再根据阿蒙顿定律求得摩擦系数.该方法可以精确地处理电子结构,并从界面电荷分布探索原子尺度摩擦行为[36−38].
势能面(potential energy surface,PES)是研究原子尺度摩擦行为的主要途径[39].特定载荷下的PES 由足够密集的势能点组成,对应相应滑移位置处的势能.构建PES 一般需要建立近700 个滑移位置的界面结构并计算其能量,涉及大量建模、计算、数据处理.本文建立了基于第一性原理方法研究二维材料层间摩擦的高通量计算程序(homogenous/heterogenous junction construction and frictional properties calculation software,HJC2S),该程序自动化建立滑移界面的原子结构,批量建立构建势能面所需计算的界面构型,高通量计算系列载荷下势能面上的所有界面结构,自动收集能量并输出摩擦性能相关数据和图像.基于该程序计算了双层氮化硼和双层石墨烯的界面层间滑移摩擦性能,研究了表面功能化对于氮化硼层间滑移摩擦力的调控.
2 二维材料层间摩擦的高通量计算程序
程序HJC2S 基于Linux 操作系统,所有计算通过基于密度泛函理论[40]的VASP (viennaabinitiosimulation package)软件包[41−43]完成.程序功能包含了自动化建模、批量产生并提交计算任务、数据提取与存储(提取并保存数据、绘制并保存图像) 3 个模块.如图1 所示,程序首先基于用户提供的二维材料结构建立同、异质结界面结构,并对界面结构进行优化;将界面一侧的材料沿着z方向按照一定间距移动,模拟系列载荷;特定界面间距下,沿着x,y方向按照一定间距移动界面一侧的材料,模拟特定载荷下沿xy平面相对滑移位置;提交对所有界面结构进行自洽计算的任务,通过作业调度系统实现计算资源的有效利用和多任务并发计算,实现二维材料界面摩擦性能的高通量计算;计算完成后,程序智能化处理并输出计算结果,用户分析数据以获得界面的纳米摩擦学性能,以下部分介绍具体的计算流程.
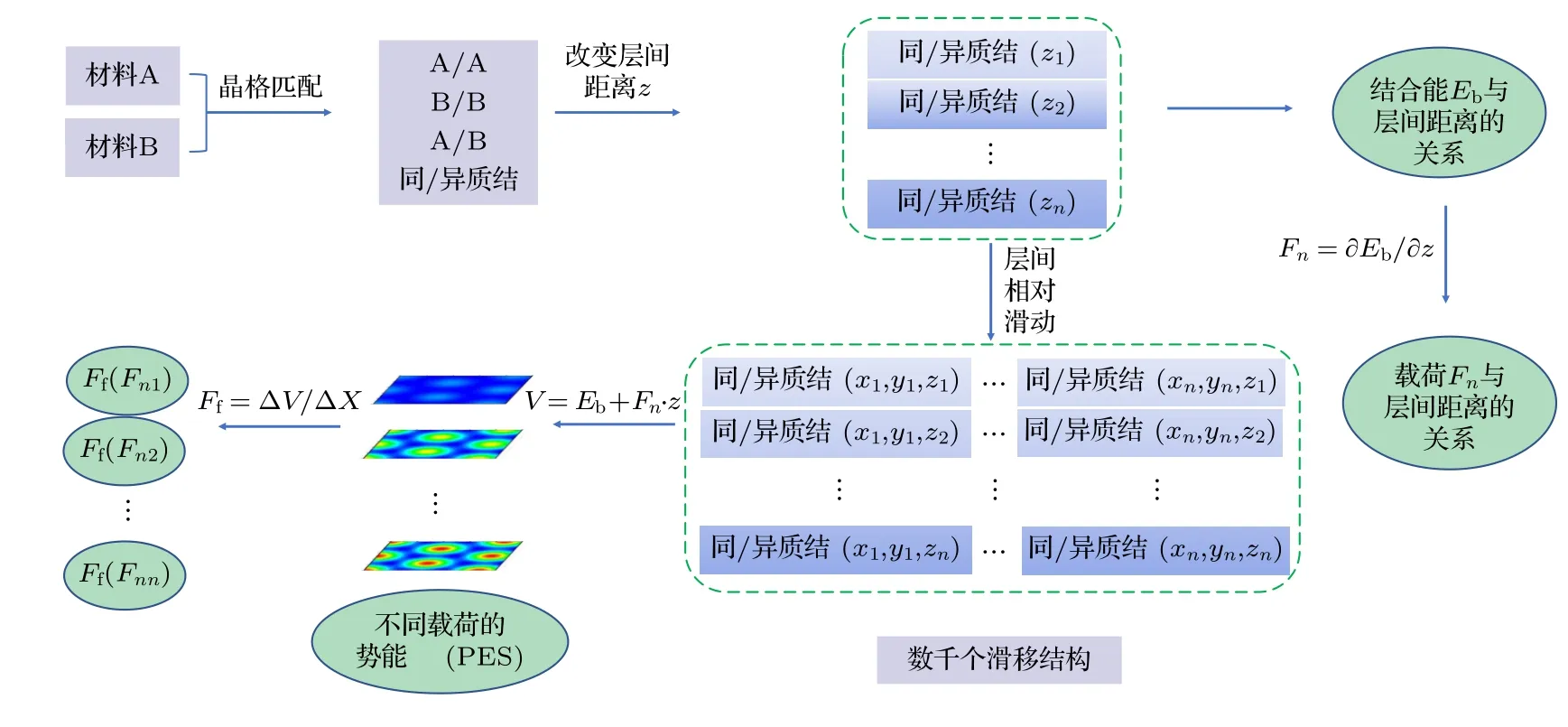
图1 二维材料界面摩擦学特性高通量计算流程示意图Fig.1.The schematic diagram showing the procedure of high-throughput calculation of the tribological property at the interface of two-dimensional materials.
2.1 构建界面结构
对于异质界面,HJC2S 程序会对提供的两种二维材料的晶胞矢量进行匹配(本文设置晶格常数失配率不超过5%,失配角不大于3°),构建异质界面结构.程序会提供各种可能晶格矢量的界面结构及该结构中界面两侧的原子数和总原子数,以供用户挑选合适的界面模型.
2.2 模拟外加载荷
HJC2S 程序通过改变界面间距模拟外加载荷,平衡间距对应零载荷.首先以平衡间距的界面结构为初始结构,设定步长并逐步减小间距,生成系列间距界面结构并对每个间距界面结构建立文件夹.然后批量提交计算系列间距界面结构的任务,并计算系列间距下界面结合能Eb.计算完成后,程序会自动提取并保存结合能以及界面间距的数据,进行数据处理并获得界面结合能随界面距离z变化的关系,通过公式求解外加载荷和界面间距之间的对应关系,自动绘制结合能随界面间距、载荷随界面间距变化的曲线图.
2.3 计算不同层间距离的势能面
HJC2S 程序通过固定界面一侧材料,使另一侧材料在xy平面做相对横向滑移来模拟界面相对滑动过程.界面被划分为M×N的网格(网格点数根据x,y方向长度设定),每一个格点位置对应相对滑移中的一个界面结构.格点划分的致密程度决定着势能面计算的精度.为了获得系列层间距下的势能面,对每一个层间距离建立文件夹,每个文件夹下有M×N个子文件夹,对应不同的滑移位置.计算完成后,程序会提取每个结构对应的滑移位置坐标和能量数据,保存并以此绘制系列层间距离下的势能面.
2.4 寻找最小能量路径
HJC2S 程序依据计算得到的势能面和最小能量路径起点和终点,采用String 方法[44]在势能面上寻找最小能量路径.String 方法是一种优秀的过渡态搜索算法,该方法以提供的起点和终点为初始值,确定两个势能极小值,然后采样多条连接两个能量极小值的曲线,通过构造曲线微分方程,使曲线逐渐逼近最小能量路径.确定最小能量路径后提取并输出路径位置坐标及能量,绘制最小能量路径及其上的静横向力曲线.
2.5 计算平均摩擦力
HJC2S 程序将筛出特定载荷下势能面上的极值点,选取邻近的最大值点和最小值点,通过两点的坐标以及能量,根据公式计算得到界面平均摩擦力,其中 ∆V=Vmax−Vmin(Vmax为势能极大值,Vmin是势能极小值),∆x是Vmax和Vmin所在位置之间的距离.程序自动计算系列外加载荷下的界面平均摩擦力〈Ff〉,输出外加载荷-平均摩擦力的对应数据并绘制外加载荷与界面平均摩擦力关系曲线.
3 计算方法
3.1 程序中涉及物理量的计算
HJC2S 程序采用如下公式计算界面结合能Eb:
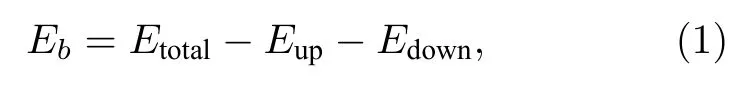
其中,Etotal是特定层间距下滑移界面的总能量,Eup,Edown分别是界面两侧材料的能量.外加载荷Fn与结合能Eb之间满足关系[45]:

程序由(1)式计算系列间距下界面结合能,由(2)式得到法向载荷与层间距的关系.
势能的计算公式如下:
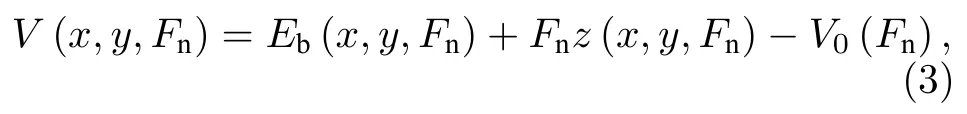
其中V(x,y,Fn)和Eb(x,y,Fn)为Fn载荷下、(x,y)位置处的势能和界面结合能,z(x,y,Fn)为Fn载荷对应的界面间距,Fnz(x,y,Fn)为抵抗外加载荷Fn所做的功,V0(Fn)是Fn载荷下势能面上的最小势能点,其计算公式如下:
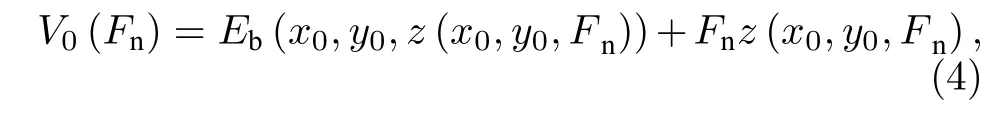
Eb(x0,y0,z(x0,y0,Fn))为零势能位置滑移体系的界面结合能,Fnz(x0,y0,Fn) 为零势能位置处抵抗外加载荷Fn的层间作用能,(x0,y0)为Fn载荷下零势能位置坐标.
特定载荷下界面平均摩擦力〈Ff〉由如下公式[45]计算:
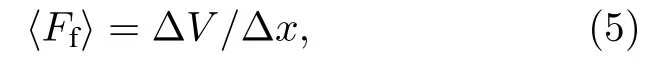
其中∆V表示该载荷下最大滑移能垒,由势能最大值与最小值决定

∆x表示势能最大值点与最小值点之间的距离.摩擦系数µ与平均摩擦力和载荷之间的关系为

3.2 本文采用的计算方法和设置
文章所有计算均采用基于密度泛函理论[40]的VASP 程序包[41−43]进行.采用广义梯度近似(general gradient approximate,GGA)的Perdew-Burke-Ernzerhof(PBE)[46]方法计算电子交换关联能,采用平面投影缀加波赝势(projector augmented wave,PAW)方法[47]描述电子和离子之间相互作用,采用范德瓦耳斯色散修正方法(density functional dispersion correction)DFT-D3 描述相邻层之间的范德瓦耳斯弱相互作用.在倒易空间中,电子波函数通过平面波基组扩展,平面波基组截断能设定为550 eV,能量和力的收敛精度分别设置为10–5eV和0.02 eV/Å.在总能计算过程中,不弛豫原子位置.布里渊区K点网格取样采用Gamma 方法,K网格大小根据具体模型由Vaspkit[48]生成.为了消除z方向上周期性结构之间的相互作用,沿垂直方向添加了20 Å厚度的真空层.
4 结果与讨论
本文采用HJC2S 程序计算了双层氮化硼(BN/BN)、双层石墨烯(Gr/Gr)、氢化氮化硼/氮化硼(H-BN/BN)和氟化氮化硼/氮化硼(F-BN/BN)的层间滑移摩擦行为.为了模拟不同载荷下的层间滑移摩擦行为,将两层之间层间距以步长0.1 Å逐步压缩,研究了层间距离为4.0—1.5 Å的Gr/Gr 和层间距离为4.0—2.5 Å的BN/BN,H-BN/BN 和F-BN/BN 的势能面、界面摩擦力和摩擦系数.
4.1 不同载荷下双层氮化硼界面摩擦力
首先研究了BN/BN 的界面摩擦行为,测试HJC2S 程序的可靠性.图2(a),(b)分别为BN/BN处于Top(T)位置和Hollow(H)位置的俯视图和侧视图.Top 位置结合能为29 meV/atom,层间距为3.39 Å,与Marom 等[49]采用PBE+Vdw 方法计算得到的BN/BN 界面层间距(3.37 Å)吻合.采用2×2 超胞模拟BN/BN 界面层间相对滑移,将Top 位置设置为初始位置,沿着xy平面滑动距离设置为5 Å × 5 Å,用于构建势能面的网格大小为0.2 Å × 0.2 Å,第一布里渊区K网格大小设为7 × 7 × 1.
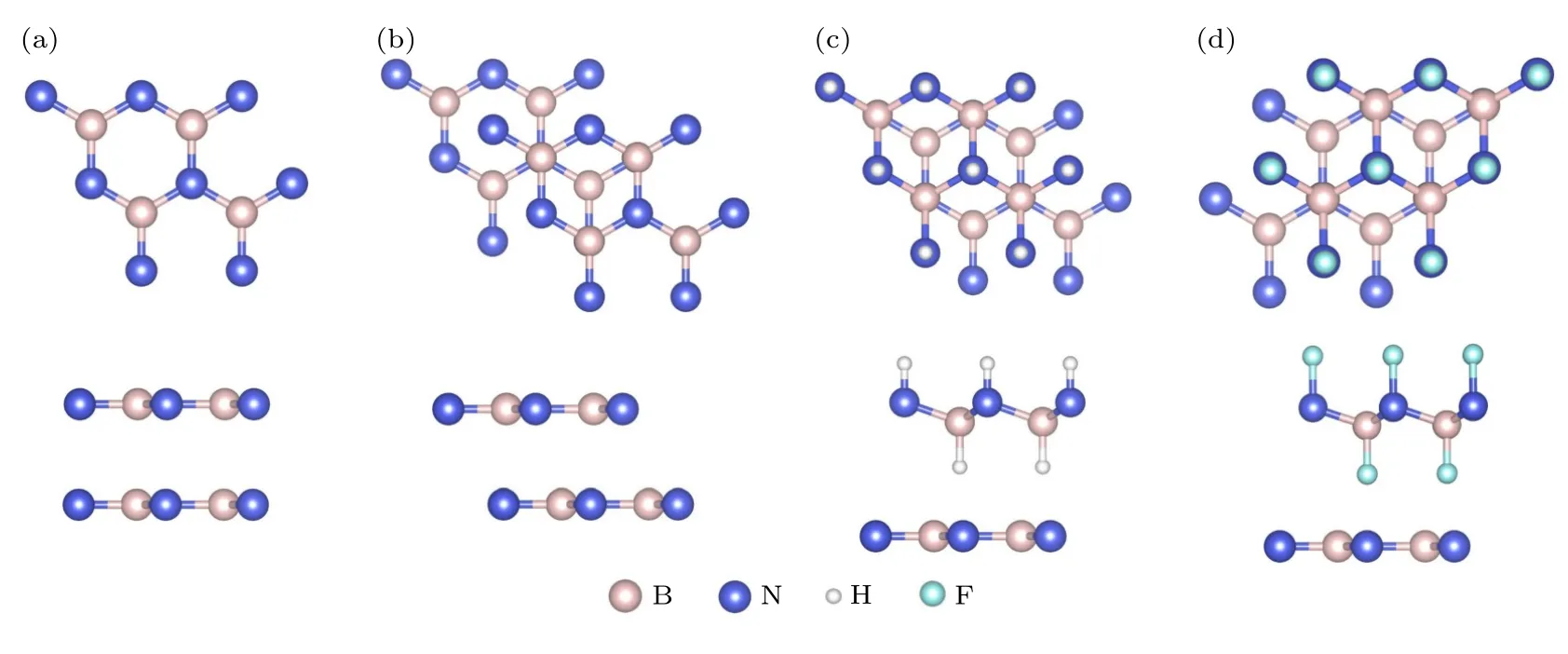
图2 BN/BN,H-BN/BN 和 F-BN/BN 双层体系原子结构的俯视图和侧视图 (a) Top 和(b) Hollow 位置BN/BN 的俯视图和侧视图;(c) H-BN/BN 和(d) F-BN/BN 在Hollow 位置的俯视图和侧视图Fig.2.Top and side views for atomic structures of BN/BN,H-BN/BN,and F-BN/BN bilayer.Top and side views of BN/BN bilayer in (a) Top and (b) Hollow positions.Top and side views of (c) H-BN/BN and (d) F-BN/BN bilayers in Hollow position.
界面结合能与层间距的关系是计算中最重要的一部分,该曲线拟合的精确度决定着计算结果的准确度.图3(a)展示了Top 位置的BN/BN 界面结合能随层间距的变化.随着层间距缩小,界面结合能逐渐变小,当层间引力达到最大值时,界面结合能达到最小值;之后,界面结合能开始增大,当界面结合能为0 eV 时,引力和斥力达到平衡;之后,斥力在层间作用中起主导作用.通过改变BN/BN 层间距来模拟载荷,根据 (2)式得到载荷与层间距的关系.如图3(b)所示,随着BN/BN 层间距减小,载荷不断增大.
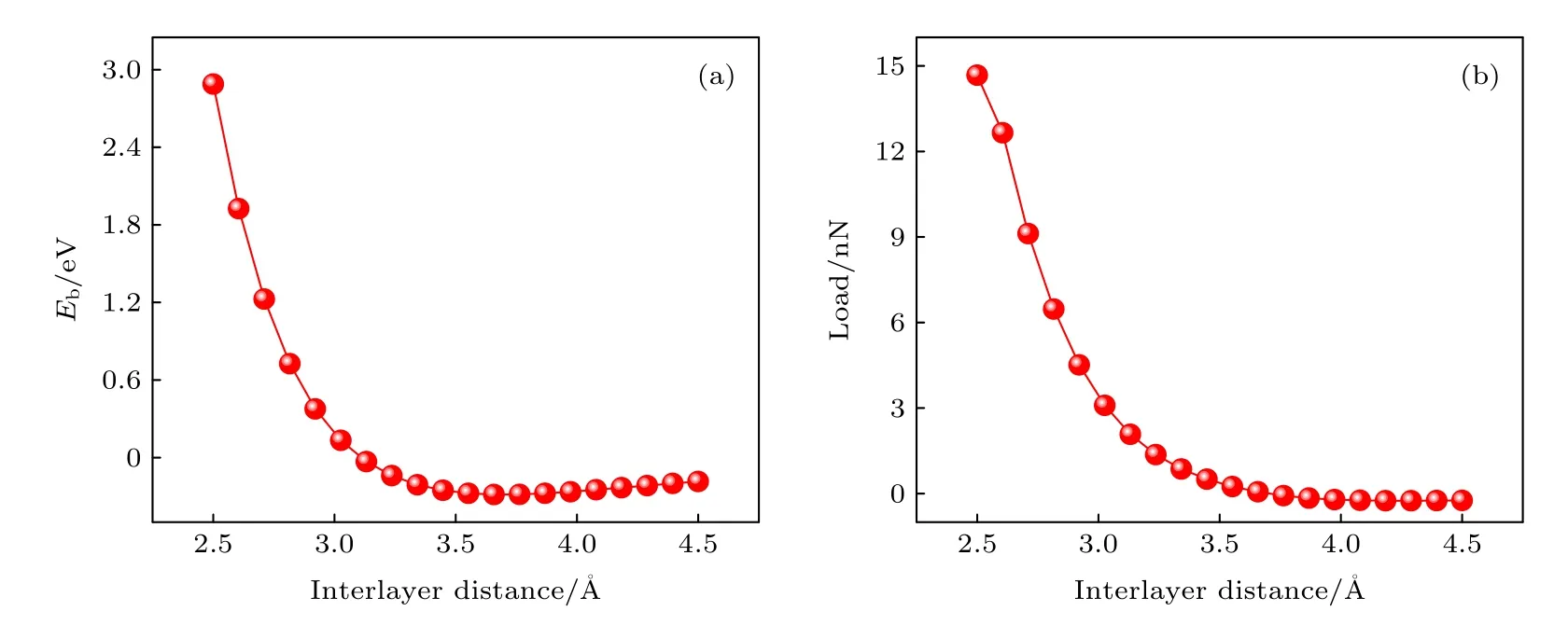
图3 BN/BN 位于Top 位置的界面结合能和载荷随层间距的变化 (a) 结合能;(b) 载荷Fig.3.The variation of interlayer binding energy (Eb) and load with interlayer distance for BN/BN bilayer at Top position: (a) Eb and (b) load.
图4(a)—(i)为0—8 nN 载荷下BN/BN 沿着xy平面分布的势能.由于BN/BN 结构本身的周期性,所以势能也呈现周期性分布.如图4 所示,0 nN 载荷下,BN/BN 的势能面比较平滑,Top 位置处势能最大,Hollow 位置处势能最小.这是因为,相较于Hollow 位置,Top 位置的正对原子更多,层间相互作用更强.随着载荷增大,抵抗负载的层间作用增大,势能面起伏逐渐增大,能垒也逐渐增大,势能最大值和最小值对应的位置并未发生变化.高势能起伏增大了滑移阻力及因摩擦耗散的能量,根据(5)式,平均摩擦力也在逐渐增大.
图4 显示了各载荷下的最小能量路径(势能面上连接势能最小值点的路径中势能起伏最小的路径),图5 为0—8 nN 载荷下最小能量路径上的能量变化曲线.从图5 可以看出,由于结构的对称性,能量随位移变化也具有周期性.沿最小能量路径进行滑移时,势能先增大后减小,然后再重复此过程.另外,随着载荷的增大,最小能量路径上的能垒逐渐增大,即界面滑移的阻力增大,从而导致更高的摩擦.
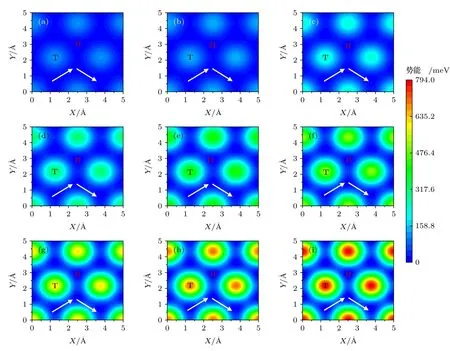
图4 不 同载 荷下BN/BN 的势 能面 (a) 0 nN;(b) 1 nN;(c) 2 nN;(d) 3 nN;(e) 4 nN;(f) 5 nN;(g) 6 nN;(h) 7 nN;(i) 8 nN.T 和H 分别表示Top 和Hollow 位置,白色带箭头线代表最小能量路径Fig.4.Potential energy surface of BN/BN bilayer under normal load of (a) 0 nN,(b) 1 nN,(c) 2 nN,(d) 3 nN,(e) 4 nN,(f) 5 nN,(g) 6 nN,(h) 7 nN and (i) 8 nN.T and H present the Top and Hollow positions,respectively.The white lines with arrow represent the minimum energy path.
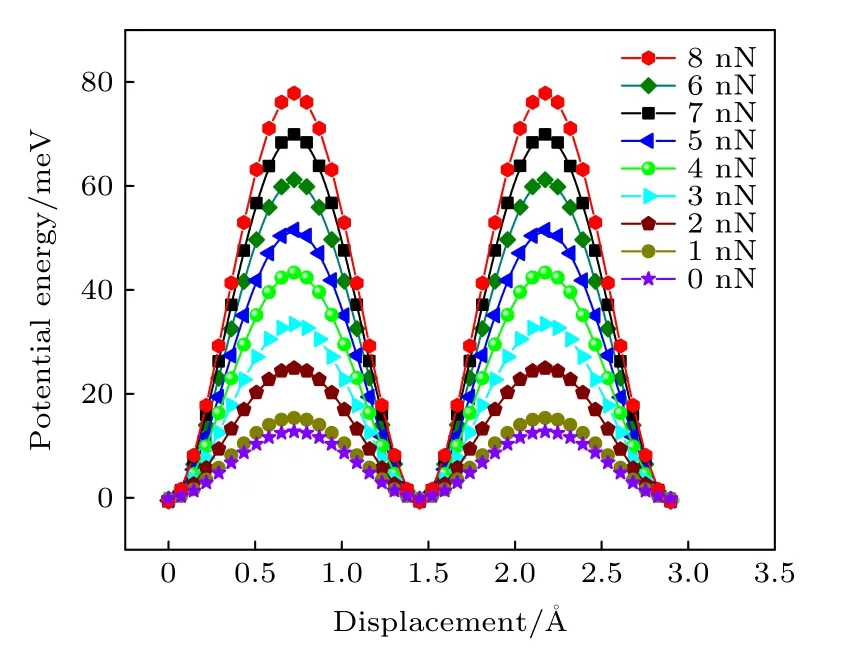
图5 0—8 nN 载荷下BN/BN 最小能量路径上势能的变化Fig.5.The variation of potential energy along the minimum energy path of BN/BN bilayer under 0–8 nN loads.
从图6(a)可以看出,随着载荷增大,BN/BN界面的平均摩擦力近似线性增大,表现出类阿蒙顿定律的特征,表明宏观摩擦力与载荷之间的关系扩展到了纳米尺度的BN/BN 滑移界面.从图6(b)可以看出,当载荷从1 nN 增大到8 nN 时,BN/BN滑移界面的摩擦系数从0.17 降至0.11.因表面结构、环境、计算方法等不同,测得的六方氮化硼界面的摩擦系数为0.025—0.7.Martin 等[50]发现超高真空下h-BN 与h-BN 界面的摩擦系数为0.7,而在潮湿空气中的摩擦系数为0.1.Wang 等[51]指出h-BN 固体润滑剂的适用温度范围为–184—538 ℃,相应的摩擦系数为0.1—0.2.An 等[23]采用DFTLDA 泛函描述BN/BN 界面的相互作用,研究了BN/BN 界面摩擦系数与载荷的变化关系,他们指出,当载荷从1 nN 增大到5 nN 时,沿着x方向滑移摩擦系数从0.1 下降至0.025,在5—10 nN 载荷下,摩擦系数从0.025 上升到0.13.本文研究了不同载荷下BN/BN 的势能面与摩擦力,摩擦系数计算结果与已有研究结果一致.
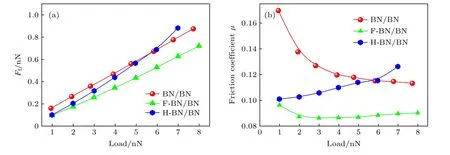
图6 BN/BN,F-BN/BN,H-BN/BN 的平均界面摩擦力(Ff)和摩擦系数(µ)随载荷的变化 (a)平均摩擦力;(b)摩擦系数Fig.6.The variation of friction force at the interface of BN/BN,F-BN/BN,and H-BN/BN bilayers with respect to the normal load: (a) Averaged interfacial friction (Ff);(b) friction coefficient (µ).
4.2 不同载荷下双层石墨烯界面摩擦力
以0.1 Å步长将层间距离从4 Å压缩到1.5 Å,计算了系列层间距下双层石墨烯(上下层均为2 个原子)之间的结合能,得到结合能随层间距变化的关系,根据(2)式得到载荷与层间距的关系,以此模拟0—13.5 nN 外加载荷.将沿xy平面滑动距离设置为2.6 Å × 2.6 Å,网格大小设为0.2 Å × 0.2 Å,第一布里渊区K网格设为12 × 12 × 1.计算了1—13.5 nN 外加载荷下Gr/Gr 的势能面,结果如图7 所示.从图7 可以发现,当外加载荷为1 nN时,势能面较为平坦,Top 位置势能最高,Hollow位置势能最低.随着载荷升高,势能最大值先增大后减小.当载荷达到12 nN 时,势能面发生了反转,即原来高势能点(Top 位置)变为低势能点,低势能点(Hollow 位置)变为高势能点,势能面趋于平坦,能垒很低.随着载荷的进一步增大,势能起伏再次逐渐增大.
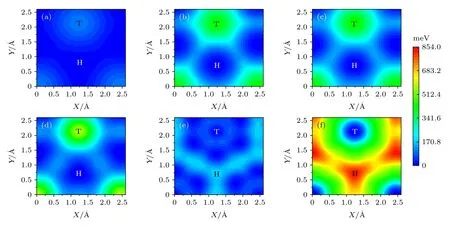
图7 不同载荷下Gr/Gr 的势能面 (a) 1 nN;(b) 4 nN;(c) 7 nN;(d) 10 nN;(e) 12 nN;(f) 13.5 nN.(a)—(d)中的H/T 对应最小/最大势能,而(e),(f)中的H/T 对应最大/最小势能Fig.7.Potential energy surface of graphene/graphene (Gr/Gr) bilayer under normal load of (a) 1 nN,(b) 4 nN,(c) 7 nN,(d) 10 nN,(e) 12 nN,and (f) 13.5 nN.T and H present the top and hollow positions,respectively.H/T in (a)–(d) is the position of minimum/maximum potential energy,whereas,T/H in (e),(f) is the position of minimum/maximum potential energy.
在势能计算的基础上,计算了1.0—13.5 nN外加载荷下Gr/Gr 界面的平均摩擦力和摩擦系数.图8(a)所示为平均摩擦力随载荷的变化曲线.如图所示,随着载荷升高,Gr/Gr 界面的平均摩擦力逐渐增大;当载荷增大到10 nN 时,平均摩擦力开始下降,直到载荷为12 nN 时,平均摩擦力达到极小值;之后,平均摩擦力随着载荷的增大再次逐渐增高.图8(b)所示为1.0—13.5 nN 外加载荷下Gr/Gr 的界面摩擦系数.从图中可以看出,随着载荷的升高,摩擦系数先减小后增大.当载荷为12 nN 时,摩擦系数最小(0.014),这是因为12 nN 下的势能面最平坦,界面滑移需要克服的能垒最低,表现出极低的摩擦系数.
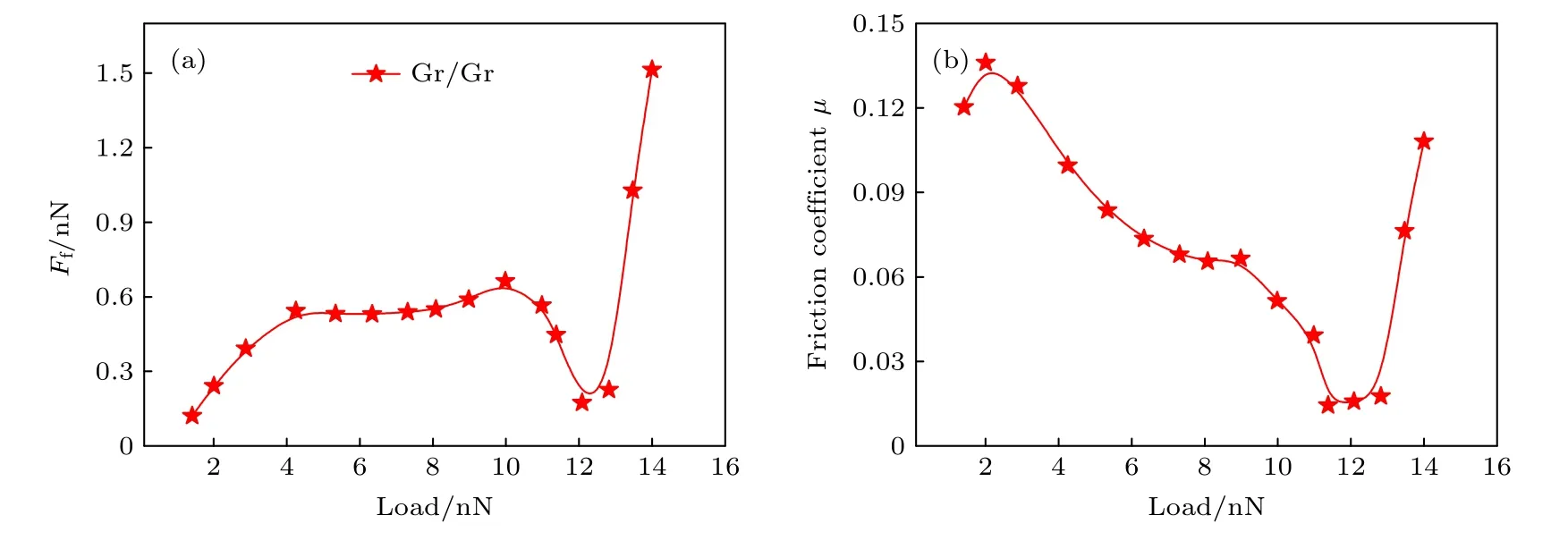
图8 Gr/Gr 界面平均摩擦力和摩擦系数随载荷的变化 (a)平均摩擦力;(b)摩擦系数Fig.8.The variation of friction force at the interface of graphene/graphene (Gr/Gr) bilayers with respect to the normal load:(a) Averaged interfacial friction and (b) friction coefficient.
本文计算的摩擦力随载荷变化的趋势成功复现了Sun 等[24]的研究结果.此外,最小摩擦系数出现的层间距为1.75 Å,与Zhang 等[52]得出的极低摩擦现象发生的层间距(1.78 Å)相近,与Sun 等[24]得出的极低摩擦现象发生的层间距(1.825 Å)略有差距,这是因为Sun 等[24]是通过固定部分原子方法计算总能,而本文采用了固定所有原子的方法计算总能,本文计算方法可以节省计算资源并减少计算时间.Fan 等[53]在实验中测得的石墨烯的摩擦系数为0.1—0.2,Wang 等[27]计算结果显示,载荷为1—9 nN 时石墨烯界面的摩擦系数为0.05—0.225.本文计算显示石墨烯界面摩擦系数为0.014—0.14,与文献[53]计算结果一致.这些均证明了HJC2S 程序和其采用算法研究界面摩擦行为的可靠性.
4.3 表面改性调控BN/BN 界面摩擦力
图2(c),(d)分别为H—BN/BN 和F—BN/BN的俯视图与侧视图.H—BN 和F—BN 中B—H,N—H,B—F,N—F 键长分别为1.20,1.03,1.36,1.44 Å.H—BN/BN 和F—BN/BN 位于Top 位置平衡结构的层间距分别为2.91 Å和3.20 Å,xy平面晶格常数为2.54 Å 和 2.57 Å.采用HJC2S程序,构建了势能面网格,大小均为5 Å × 5 Å,网格尺寸为0.2 Å × 0.2 Å,计算了0—8 nN 载荷下H—BN/BN 和F—BN/BN 的势能面及界面平均摩擦力和摩擦系数.
图6(a)为BN/BN,F—BN/BN 和H—BN/BN滑移界面平均摩擦力随载荷变化的曲线,如图所示,1—8 nN 载荷下,3 种滑移界面平均摩擦力均随载荷增大而增大.图6(b)所示为3 种滑移界面摩擦系数随载荷变化的曲线.随着载荷增大,H—BN/BN 界面摩擦系数增大,F—BN/BN 界面摩擦系数先减小后缓慢升高,摩擦系数值为0.08—0.10.当载荷小于6 nN 时,H—BN/BN 滑移界面的摩擦系数小于BN/BN 滑移界面的摩擦系数;当载荷大于6 nN 时,H—BN/BN 滑移界面的摩擦系数大于BN/BN 滑移界面的摩擦系数.可以明显地看出F—BN/BN 滑移界面的平均摩擦力和摩擦系数均小于BN/BN 和H—BN/BN,表明表面氟化显著降低了摩擦.
电荷转移是影响摩擦力的主要因素[54−56],为了探析氟化降低BN 界面摩擦的内在原因,计算了BN/BN 以及F-BN/BN 的电荷分布.图9为0 nN和8 nN载荷下BN/BN 和F-BN/BN在势能最高位置和最低位置处的差分电荷密度图.由于F 原子的电负性较强,F-BN/BN 层间电子聚集在F 原子附近,层间电荷转移较小;BN/BN 层内电子结合能力较弱,层间电荷转移较为明显.从图9可以看出,0 nN 和8 nN 载荷下,BN/BN 由Top位置(势能最大值点)滑移到Hollow 位置(势能最小值点)时,电荷分布变化显著.氟化后BN 界面从高势能点滑移到低势能点位置时,电荷分布无明显变化,所以滑移过程中的能垒较低,层间摩擦小,因此降低了BN 界面摩擦.
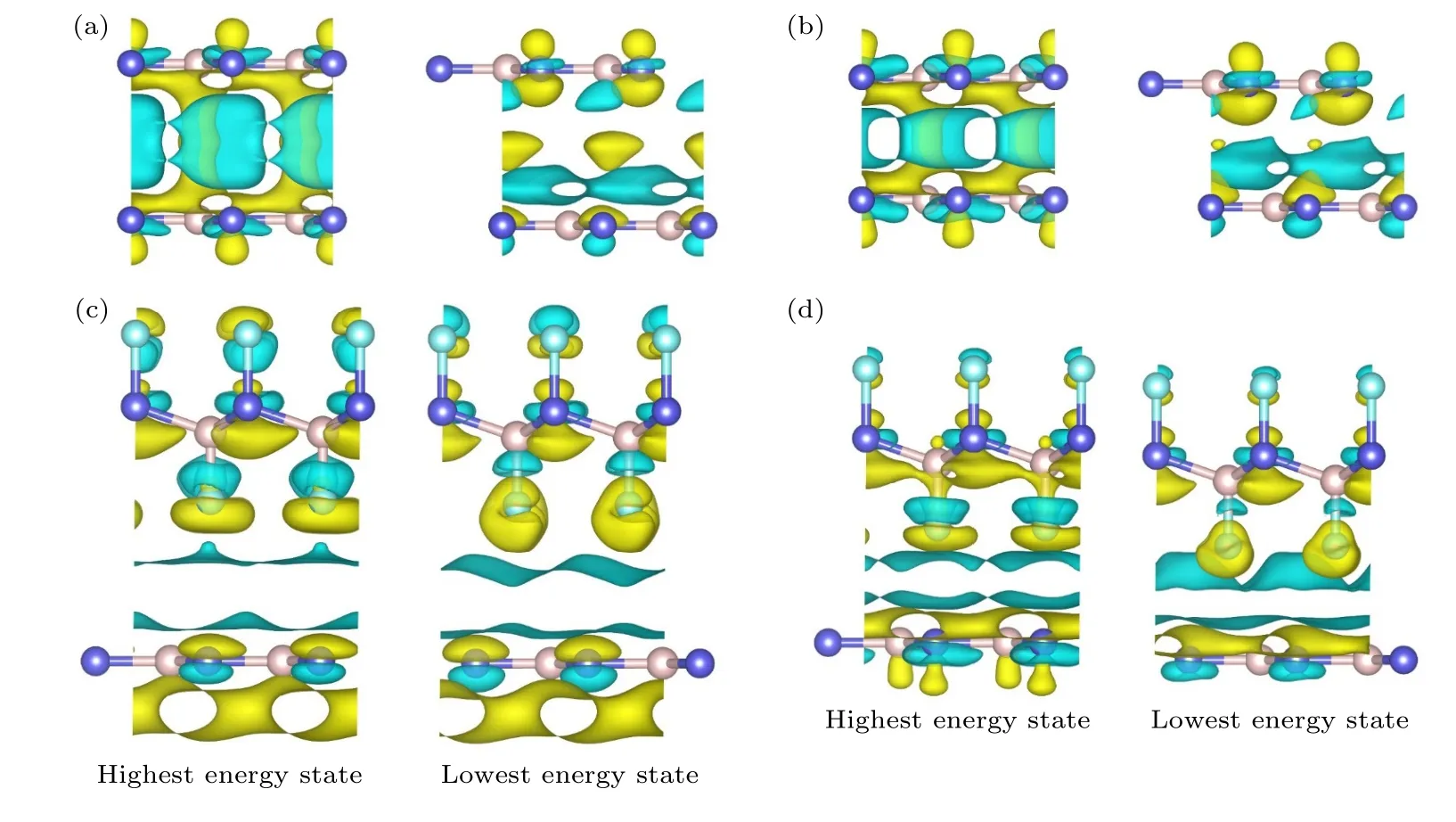
图9 BN/BN 和F-BN/BN 的最高和最低势能态在0 nN 和8 nN 载荷下的差分电荷密度 (a) BN/BN,0 nN;(b) BN/BN,8 nN;(c) F-BN/BN,0 nN;(d) F-BN/BN,8 nN.黄色代表电子聚集,蓝色代表电子损失;左边和右边的图分别对应最高和最低势能态;图(a)和(c)中的等值面设置为0.00006e/Bohr3;图 (b) 和 (d) 中的等值面设置为0.0003e/Bohr3Fig.9.Differential charge density of BN/BN and F-BN/BN bilayer at the highest and lowest potential energy state under 0 nN and 8 nN loads.BN/BN bilayer under (a) 0 nN and (b) 8 nN load,F-BN/BN bilayer under (c) 0 nN and (d) 8 nN loads.The yellow color represents electron aggregation,and the blue color represents electron dissipation.The isosurface value in figure (a) and (c) is set to 0.00006e/Bohr3;the isosurface value in figure (b) and (d) is set to 0.0003e/Bohr3.
4.4 方法的适用性与局限性
DFT 方法可以深入到量子尺度,从电子分布的角度解释摩擦现象.尽管该方法已经被广泛的用来预测层间滑移的摩擦系数,但该方法仍存在一些局限性.首先,在层间相对滑动的过程中,本文将上下层均设为刚性,并未考虑层间弯曲的影响.其次,本文通过改变计算体系的堆叠构型以模拟层间滑动,因此计算得到的实际结果本质是静摩擦,而非动摩擦.另外,DFT 计算本身具有局限性,如在计算过程中无法直接引入温度,因此该方法并未考虑温度的影响;计算通常是对数十个原子进行,无法对几百个原子进行计算.
5 结论
报道了一个基于第一性原理方法计算二维材料层间滑移摩擦行为的高通量、自动化计算程序.该程序实现了同、异质结界面自动化建模、计算任务自动生成及提交、智能提取并存储计算结果、自动处理数据并计算摩擦力和摩擦系数,极大地缩短了建模和处理数据的时间.采用此程序模拟计算了外加载荷下双层氮化硼层间滑移的摩擦,得到的摩擦系数与实验和已有计算结果一致,同时研究了双层石墨烯层间滑移的摩擦力和摩擦系数,计算结果与文献结果相吻合,证明了该程序计算结果的可靠性.此外,我们研究了表面改性对双层氮化硼层间滑移摩擦的影响,结果表明表面氟化可以很好地降低摩擦,提供了降低六方氮化硼界面摩擦的表面改性方法.
