新生儿糖尿病6例病例特点及临床分析
2023-02-01王淑琴高怡青朱磊侯梦刘金凤薛颖
王淑琴,高怡青,朱磊,侯梦,刘金凤,薛颖
新生儿糖尿病(neonatal diabetes mellitus,NDM)多在6月内发病,少数可发生于1岁前,病因以单基因突变为主[1]。随着对NDM认识的提高,发病率上升,最近研究显示其发病率为 1/9 000~1/160 000[2]。NDM按病程分为永久性新生儿糖尿病(permanent neonatal diabetes mellitus,PNDM)和暂时性新生儿糖尿病(transient neonatal diabetes mellitus,TNDM),以NDM发病的综合征一般归类为PNDM。PNDM可伴有部分或完全的胰岛素缺乏,诊断时临床表现包括宫内生长迟缓、高血糖、糖尿、渗透性多尿、糖尿病酮症(diabetic ketosis,DK)、糖尿病酮症酸中毒(diabetic ketoacidosis,DKA)等,目前已明确的基因突变包括ABCC8、GCK、INS、KCNJ11、PDX1等,病程因基因型不同而各不相同[3]。TNDM一般在发病后18个月内缓解,染色体6q24差异性甲基化异常是TNDM常见病因[4],约半数以上的TNDM在儿童期或青春期复发,复发后可予磺脲类药物或较小剂量的胰岛素治疗,优选治疗方案仍需进一步探讨[5]。TNDM诊断时较少发生DKA,所需胰岛素治疗剂量低。此外,以NDM发病的综合征如Wolcott-Rallison综合征、IPEX综合征、Wolfram综合征等,对糖尿病治疗仍以胰岛素治疗为主,伴发症状不同预后亦不同。对于新生儿期起病的NDM很难直接诊断PNDM、TNDM,在随访过程中根据治疗情况而定。明确基因的致病性变异对于临床治疗重要,多数携带KCNJ11或ABCC8致病性变异的病人可使用磺脲类药物治疗。使用磺脲类药物治疗不仅可以改善血糖控制,亦可改善病儿的神经认知功能[6-7]。本研究通过对6例NDM临床特点及随访情况进行分析,为临床诊疗提供依据。
1 资料与方法
1.1 一般资料选择2013年2月至2021年6月于徐州医科大学附属徐州儿童医院就诊的6例NDM为研究对象,收集临床资料进行分析。本研究已获得徐州医科大学附属徐州儿童医院医学伦理委员会批准(伦2021-06-01-K05)。
1.1.1 诊断标准 6月内婴儿持续高血糖(血浆葡萄糖浓度150~200 mg/dL),同时排除应激、感染、药物等引起的一过性高血糖,对于1岁内发病经基因检测证实亦可诊断为NDM。
1.1.2 诊断和治疗方法 收集6例NDM病儿临床资料,包括:出生史、出生体质量、性别、C肽、糖化血红蛋白(HbA1C)、抗胰岛细胞抗体(ICA)、抗胰岛素抗体(IAA)、抗谷氨酸脱羧酶抗体(GADA)、静脉血葡萄糖、血气分析、血电解质及尿酮等。6例病儿均住院治疗。合并糖尿病酮症酸中毒(DKA)或糖尿病酮症(DK)者采用补液及小剂量胰岛素纠酮体治疗。DKA、DK纠正后采用胰岛素皮下注射方案。5例病儿经用格列本脲口服替代治疗,用量从0.1 mg·kg-1·d-1, 1至 2 周内逐渐增加至 0.8 mg·kg-1·d-1,同时逐渐减少胰岛素用量直至停用胰岛素。如停胰岛素后,格列本脲治疗可平稳控制血糖,提示治疗有效。如格列本脲增至0.8 mg·kg-1·d-1,仍不能减少胰岛素用量,提示格列本脲治疗无效。
C肽测定采用化学发光法(瑞士罗氏公司),ICA、IAA、GADA测定采用酶联免疫吸附试验(瑞士罗氏公司),HbA1C测定采用高效液相色谱法。
基因检测方法:病儿近亲属知情同意后,根据徐州医科大学附属儿童医院基因检测相关流程,抽取血标本(病人及父母静脉血各 2 mL)送至第三方进行检测。病例 1、2、3、4、6均送至南京金域医学检验,病例5送至康圣环球海斯特。南京金域医学检验公司通过Ion-Torrent-PGM技术对新生儿糖尿病相关基因进行测序。康圣环球海斯特使用对应试剂盒提取样本 DNA 后,使用探针(GenCap)捕获相关候选基因进行高通量测序。
2 结果
2.1 一般资料6例NDM男孩5例,女孩1例。合并小于胎龄儿3例,病儿诊断年龄为3~256 d,中位年龄为50 d。随访时间4.5~105.2个月。1例因病儿诊断NDM后发现其母亲为空腹血糖受损,余病例无糖尿病家族史。见表1。
2.2 初次诊断时情况3例以发热感染为首发症状,3例以反应差起病。初诊时4例合并糖尿病酮症酸中毒,1例合并糖尿病酮症。诊断时平均糖化血红蛋白 11.4%(7.35%~13.8%),C肽均降低,ICA、IAA、GADA抗体均阴性。急性期均用胰岛素治疗,血糖控制良好。5例病儿行格列本脲经验性治疗,4例有效,1例无效停药,成功率为80%,均无明显不良副作用。见表1。
2.3 随访情况随访时间最短4.5月,最长8年6月。1例TNDM,4例PNDM,1例随访4.5月并发肝功能衰竭死亡,因随访时间短未分型。6例行基因检测,KCNJ11基因突变4例,其中1例为首次报道。1例TNDM急性期予胰岛素治疗后,转为格列本脲治疗成功,病儿6月龄时停格列本脲,8岁时糖尿病复发,予长效小剂量胰岛素治疗血糖可控制平稳。3例使用格列本脲PNDM随访至今血糖均控制良好,无明显不良反应,经基因检测证实均为KCNJ11突变所致。1例使用胰岛素治疗PNDM,随访至今1岁4月,血糖波动相对较大。5例生长发育与同性别同龄儿相仿,无语言运动发育落后。见表1。
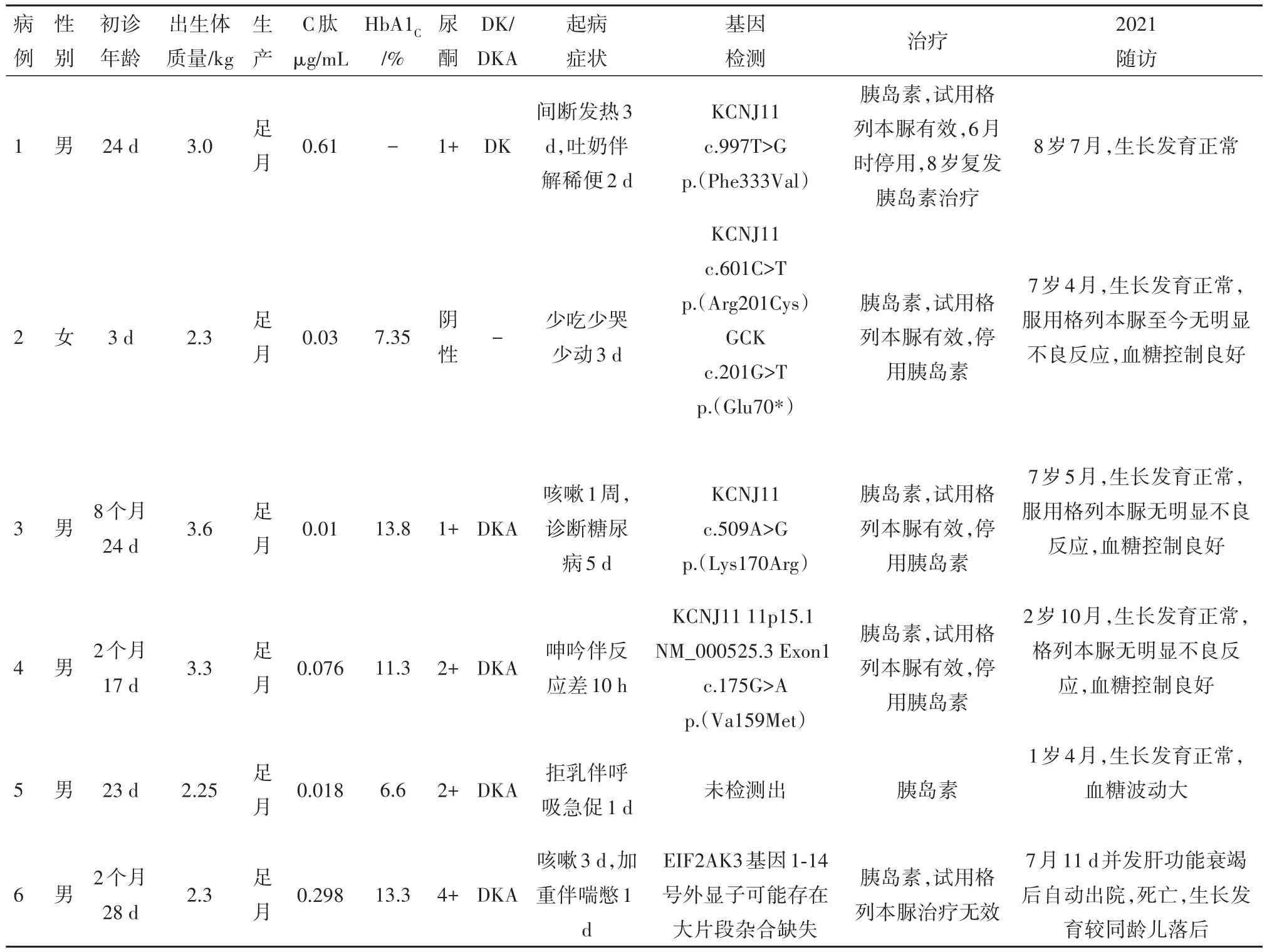
表1 6例新生儿糖尿病临床特点分析
3 讨论
新生儿糖尿病多指小于6个月婴儿的持续性高血糖(血浆葡萄糖浓度 150~200 mg/dL)。然而最近的研究发现,单基因形式的NDM在1岁时仍可发病[5,8],因此推荐1岁以下发病的糖尿病病人行基因检测以精准化指导诊疗[9]。单基因NDM的预后和治疗方案在很大程度上取决于哪些基因受到影响。基因检测使NDM精准化治疗比例得到很大提高[10]。正如本文中病例3,初诊年龄8月余,发病年龄超过传统关于NDM认知6月龄内,行基因检测明确为KCNJ11突变,行格列本脲治疗有效,停用胰岛素,服用格列本脲控制血糖良好,无明显低血糖事件发生,已随访至7岁5月,病儿生长发育正常,此例病儿精准化治疗得益于基因检测。
TNDM的儿童通常在婴儿期(13~18周龄)高血糖得到缓解。然而,在青春期或成年期可能会复发[11]。TNDM最常见的原因6q24基因座上的基因过度表达,由于6号染色体单亲二倍体、该区域的异常复制(父系复制)或DNA甲基化的丢失,从而激活母体等位基因而导致6q24处印记缺失的结果[12]。第二个最常见原因是编码电压依赖性钾通道亚单位(KATP)的两个基因突变[13],分别为KCNJ11编码的Kir6.2亚单位,ABCC8编码SUR1亚单位[14],这两个基因中的任何一个激活突变都会导致KATP通道不适当地开放,从而导致尽管高血糖但细胞膜无法去极化和胰岛素释放障碍。
本研究中病例1为TNDM病儿,以发热感染就诊,合并糖尿病酮症,胰岛素治疗病情稳定后,转换为格列本脲并停用胰岛素治疗,血糖控制良好。6月大时病儿病情缓解,停用格列本脲,本例病儿缓解期较文献报道晚,但在随访过程至8岁时糖尿病复发,调整为小剂量胰岛素治疗。此例病儿基因检测为KCNJ11杂合突变,Phe333Val突变未见任何文献报道,生物信息学认为其对KCNJ11基因功能存在一定影响,结合父母均未检出Phe333Val突变,结合诊疗随访情况,其突变考虑极可能为新发突变位点导致TNDM。
PNDM最常见的原因是激活KCNJ11或ABCC8的杂合突变[15-16],占所有新生儿糖尿病病例50%以上。这两种基因突变也是TNDM的第二位常见原因。这两个基因突变的病人对磺脲类药物治疗敏感[17]。大约25%的ABCC8或KCNJ11基因突变病人患有神经认知功能障碍,从精神运动障碍到与严重癫痫相关的认知发育迟缓[18],使用磺胺类药物治疗使HbA1C正常化,同时减少ABCC8或KCNJ11突变的新生儿糖尿病中低血糖的发生率,亦可改善神经认知功能[19]。继KCNJ11、ABCC8基因突变之后,第二种最常见的永久性新生儿糖尿病是胰岛素基因INS突变。INS突变可出现在20%的PNDM婴儿中。该基因突变导致胰岛素蛋白错误折叠,从而导致内质网应激增加和最终β细胞死亡[20]。 INS突变病人的诊断时间晚于ATP敏感性钾离子通道突变携带者(11周比 8周)[8]。除永久性新生儿糖尿病外,病人没有任何其他表型特征,需要终身胰岛素治疗[21]。
本研究中1例经基因检测未发现明确致病基因的PNDM,故未予格列本脲转换治疗,诊断后予胰岛素治疗至今,生长发育正常,但血糖控制因年龄小波动相对较大。另3例予格列本脲治疗有效PNDM病儿,经基因检测证实均为KCNJ11基因突变。此3例病儿随访至今,生长发育正常,目前关于格列本脲长期服用相关疗效及副作用报道极少,本组病例中最长已服用7年4个月,可平稳控制血糖,且无明显不良反应,提示格列本脲相对较安全可靠。
KCNJ11基因突变3例病儿中有1例合并GCK杂合突变,该病儿因“少吃少哭少动3 d”入院就诊,入院后结合基因检测KCNJ11基因突变诊断为新生儿糖尿病,格列本脲治疗有效,此外该病儿合并GCK基因杂合突变,可导致MODY2发生,基因检测证实GCK基因突变来源母亲,后母亲监测血糖发现空腹血糖高,无临床症状,目前经饮食干预及生活锻炼后糖化血红蛋白可维持正常范围,提示该病儿未来亦可能合并MODY2发生,目前仍需继续随访该病儿病情进展情况。
KCNJ11突变病人可能存在广泛的神经认知障碍,睡眠障碍,注意缺陷多动障碍以及行为发育延迟[22]。本研究中3例病儿经诊断后及时予格列本脲治疗,随访至今未发现严重低血糖事件,生长发育与同性别同龄儿相仿,提示格列本脲可平稳控制KCNJ11突变导致NDM病儿的血糖,同时可能对病儿神经认知发育有改善作用,但本研究长期使用格列本脲治疗KCNJ11突变导致的NDM仅3例,未来仍需更多相关的病例纳入研究。这也强调对于KCNJ11突变导致的NDM病儿中,格列本脲比胰岛素治疗不仅更好的控制病儿血糖,并可改善神经认知功能,建议NDM诊断后尽早行基因检测以提供精准化治疗。
NDM还包括与NDM相关的综合征,一般归类于PNDM,发病机制包括胰岛β细胞破坏、胰腺发育不全或再生障碍、胰岛β细胞功能受损或严重胰岛素抵抗等[1]。最常见的综合征是Wolcott-Rallison综合征(Wolcott-Rallison synkdrome,WRS),是一种由EIF2A突变引起的常染色体隐性疾病,EIF2A是一种编码翻译起始因子2-α激酶3的基因,在内质网的调节中起重要作用。其他特征是肝功能障碍和骨骼发育不良[23]。
任何患有与骨骼发育不良和、或急性肝衰竭有关的PNDM,都应怀疑患有 WRS[24]。由于该综合征的主要表现特征可能发生在疾病过程的后期,因此,在任何新生儿或6个月之前发生糖尿病的婴儿中,应怀疑患有WRS。其临床过程非常多变,糖尿病发病初期,骨发育不良和反复肝衰竭是病人最典型的特征[25]。通常,糖尿病发生在6个月之前,骨发育不良在一两岁之前。肝衰竭可能发生在疾病过程中的任何时间,并可能在糖尿病发病后首次出现,由于病人年龄小,一些临床表现可能缺失,在进行 WRS 的完整诊断之前可能会发生死亡。本研究中病例6,2个月28 d时以“咳嗽3 d,加重伴喘憋1 d”入院,入院后完善检查示糖尿病酮症酸中毒,予胰岛素治疗病情稳定后,试用格列本脲治疗无效,基因检测示EIFIA3基因1~14号外显子可能存在大片段杂合缺失。随访中病儿合并3次肺部感染,7个月11 d时并发肝功能衰竭后自动出院死亡,此例病儿先发生NDM,随后出现肝功能衰竭,未发现骨发育不良,考虑病程短,仍应考虑WRS可能。
综上所述,1岁内发病糖尿病病人应考虑基因检测,因为NDM的临床表型与基因型异质性密切相关。磺胺类药物可以用于治疗 KATP通道基因突变(KCNJ11和ABCC8),并可改善神经认知功能。建议所有新生儿糖尿病病人进行密切随访,并关注磺脲类药物长期治疗反应。
