家蚕品种桂蚕8号亲本全基因组重测序研究初报
2023-01-28黄文功浦月霞刘艳伟苏红梅韦博尤安春梅谭福洋王平阳闭立辉
黄文功,浦月霞,刘艳伟,苏红梅,韦博尤,安春梅,谭福洋,王平阳,闭立辉
2020年广西蚕业技术推广站育成抗血液型脓病新品种——桂蚕8 号[1],4 个杂交亲本“锦”(9MN)“绣”(芙H)“壮”(7MH)“丽”(87H)均为连续添食BmNPV病原筛选固定而形成,抗血液型脓病特征明显,综合性状优良。为研究4个亲本基因组水平的特征,本研究抽样并委托深圳华大基因股份有限公司开展全基因组重测序,将测序文库比对到家蚕参考基因组上,评估比对质量,开展单核苷酸多态性(SNP)、插入缺失(Indel)、结构性变异(SV)、拷贝数变异(CNV)等分析,为下一步挖掘优异基因做好前期工作。
1 分析方法
1.1 全基因组重测序建库测序和原始数据过滤方法
采用BGISEQ 平台构建DNA 小片段文库,片段读取长度为150 bp,每个样本的采集数据量为20 G,用SOAPnuke[2]对数据进行接头、低质量reads、杂质数据和去N过滤后,最终得到高质量的可用数据CleanData,过滤参数为“filter-n0.1-l20-q0.5-Q2-G”。
1.2 比对方法
应用BWA[3-4]的“mem-t 5-M”算法,将CleanData比对到西南大学研发的参考基因组SilkDB 3.0(https://silkdb.bioinfotoolkits.net)上,生成比对结果文件sam 文件,用samtools 的sort 工具将sam 文件转变为排序后bam 文件,用qualimap分析比对质量,再用samtools软件过滤比对质量小于30的reads。
1.3 SNP和InDel检测方法
应用GATK 软件开展SNPs 和InDels 检测,用Annovar软件对SNPs和InDels进行注释和统计[5],检测的过程如下。
(1)利用Picard的Mark Duplicate工具标记比对结果中的duplication数据,避免PCR-duplication对后续变异检测结果的影响。
(2)使用GATK的HaplotypeCaller工具进行检测获得所有位点的GVCF文件,使用GenotypeGVCFs工具进行变异检测SNP和Indel变异位点,再进行过滤。
(3)使用bcftools[6]拆分得到单个样品的变异结果。
(4)基于基因组的gtf 文件分别对每个样品的SNP和Indel位点进行Annovar[5]注释。
1.4 SV检测方法
应用BreakDancer[7]检测SV,检测参数为“-q 35”,过滤参数为“-m 100 -x 1000000 -s 30 -d 5”。
1.5 CNV检测方法
使用Control-FREEC(v11.5)[8]软件检测样品里面的拷贝数变异(CNV)区域。
2 结果与分析
2.1 重测序数据产出与质量
单个品种测序数据经过滤后的Clean reads 约2亿条,碱基数量近300亿bp,GC含量在37.90%左右,比参考基因组GC含量值(38.32%)低;Q20在95.73%~96.48%之间,Q30在90.61%~92.20%之间,重测序数据质量可靠。
2.2 比对结果分析
选用的参考基因组(SilkDB 3.0)大小为454 710 009 bp,统计比对到参考基因组上各染色体区域的覆盖度和测序深度分布情况,桂蚕8号4个亲本reads比对率均接近99%,平均测序深度介于53.25×至66.65×之间,覆盖深度从1×至51×区域的百分比介于96.09%~62.82%之间,参考基因组被均匀覆盖,随机性良好,详见表1、图1。
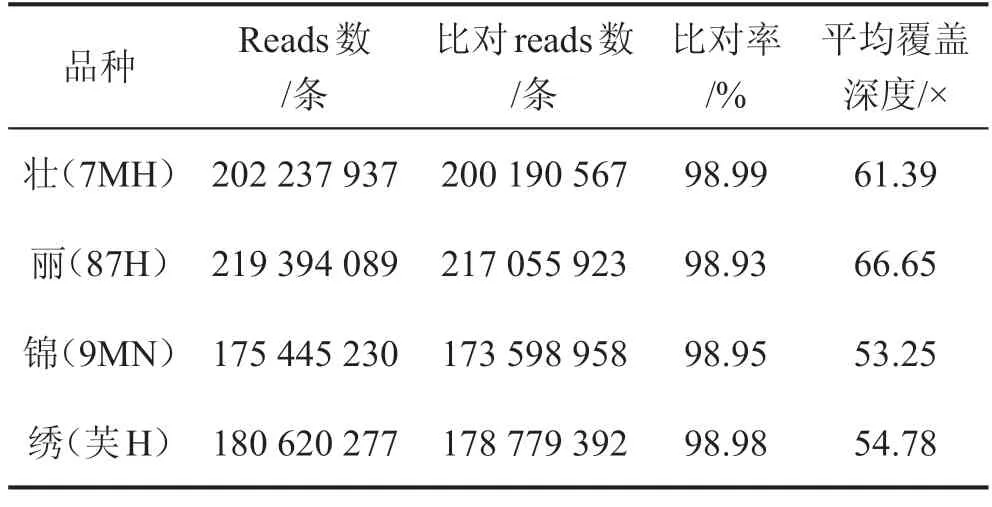
表1 比对质量情况
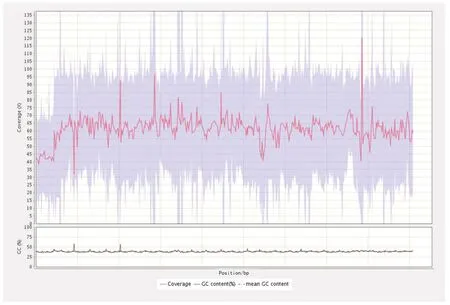
图1 参考基因组的覆盖分布图
2.3 单核苷酸多态性(SNP)分析
单核苷酸多态性(Single Nucleotide Polymorphisms,SNPs)是由单个核苷酸改变引起的染色体基因组的多样性,包括单个碱基的转换(transition,Ts)和颠换(transversion,Tv)。本研究采用GATK 软件进行多样本call 变异,得到所有样品的SNP 基因型信息,经过滤后挑选每个样品的位点信息,去掉缺失和与参考基因组一致的位点后统计每个样品的SNP 总数,包括杂合SNP、纯合SNP、转换(Ts)、颠换(Tv)的数量,4 个杂交亲本SNP 总数均超过600 万,纯合SNP比例均为98.09%,杂合SNP占比较低,转换(Ts)与颠换(Tv)的比值均为1.27左右,详见表2。

表2 SNP数量及类型信息统计
对SNP变异类型进行汇总比较,4个品种同类型碱基变异丰度基本一致,均为A 与G、C 与T 之间碱基转换(Ts)发生位点较高,A 与T、A 与C、G 与C、G与T之间碱基颠换(Tv)较低,详见图2。

图2 各种类型碱基突变丰度柱形图
将得到的每个变异位点所在的基因信息注释后,统计染色体基因的上下游区域、外显子、内含子、基因间区中变异位点的数量,同时判断位于外显子区域的SNP 突变是否会导致蛋白质序列发生变化,即突变是同义突变还是非同义突变(见表3)。4个亲本SNP 在上下游区域、外显子、内含子、基因间区等区域数据规律基本一致,大多数SNP 突变位于基因间区和内含子区域,位于这2个区域的突变大多数属于无义突变,一般不会对基因表达产生影响。位于基因上游区域的SNP位点处于16万个左右。位于终止密码子区域的SNP 突变位点低于1 000 个,会导致形成终止密码子的变异位点数量比引起终止密码子消失SNP 多。在外显子区域,引进同义突变的SNP 均超过10 万个,引起非同义突变的有均超过3.7 万个。在可变剪切位点、基因下游等位置亦有大量SNP分布。

表3 SNP突变注释信息统计表
2.4 插入缺失(InDel)分析
InDel 是指在DNA 上发生的核苷酸或核苷酸序列的插入和缺失,采用GATK 检测InDel 位点信息,先得到所有样品的基因型信息(genotype),再从中挑选每个样品的InDel 信息,经过滤后统计样品的InDel 总数、杂合Indel、纯合InDel 的数量,去掉缺失和与参考基因组一致的位点后,得到单个样品的InDel 位点,然后统计每个样品的InDel 总数,杂合Indel、纯合InDel 的数量。每个亲本的Indel 总数约140万,其中插入位点为66万~68万,缺失位点为74万~75万,纯合位点约占插入位点的92%(表4)。

表4 InDel数量统计
对InDel的长度信息进行统计,得到突变频率较高的长度为1~20 bp的InDel数量分布图,从图中可看出,4 个品种的插入和缺失长度与数量趋势一致,插入和缺失长度主要集中在长度为1~10 bp,随着InDel长度的增加,数量越少(图3)。

图3 InDel长度分布图
基因组注释得到每个基因位置信息,使用Annovar注释,统计基因的上下游区域、外显子、内含子、基因间区中变异位点的数量,同时判断InDel 变异会否导致移码突变。Indel分布趋势与SNP基本一致,但未见位于上游/下游区域的Indel(表5)。

表5 InDel突变位点注释信息统计
3.6 结构性变异(SV)检测分析
本研究中应用BreakDancer 检测结构性变异(Structural variation,SV)数量和总长度情况,包括长度在50 bp 以上的长片段序列的插入(INS)、缺失(DEL)、倒位(INV)、染色体内易位(ITX)和染色体间易位(CTX)等,桂蚕8号的4个亲本均有一定数量的结构性变异(表6、表7)。4个亲本均为缺失位点最多,插入位点最少,从相同类型的结构性变异来看,日系亲本“壮”(7MH)和“丽”(87H)插入位点比2个中系亲本多,缺失位点比中系亲本少,倒位数量相当,染色体内易位和染色体间易位略多于中系亲本。

表6 各类型结构性变异(SV)数量统计

表7 各种结构性变异(SV)总长度统计
从结构性变异总长度来看,插入总长度亦为日系亲本长;缺失长度相当,但日系亲本缺失数量较少,单个缺失位点长度较长;染色体内易位(ITX)单位点平均长度远大于染色体间易位(CTX)。
基于基因组注释得到每个基因位置信息的gtf文件,使用Annovar注释软件将变异位点所在的基因信息注释出来,并统计基因的上下游区域、外显子、内含子、基因间区中结构性变异位点的数量,结构性变异主要分布在基因间区、内含子、外显子、基因上游、基因下游,在可变剪切位点、UTR5、UTR3 及位于一个基因的上游同时也是另一个基因的下游区间亦有分布。

表8 结构性变异(SV)位置统计
3.7 拷贝数变异(CNV)检测分析
拷贝数变异(CNV)一般指长度为1 kb 以上的基因组大片段的拷贝数增加或者减少,依靠特定位置的测序深度与平均测序深度来估算的。本研究使用Control-FREEC 软件来检测每个样品的CNV变异,根据设定的家蚕染色体倍性为2,滑窗长度为50 000 bp,自动计算固定长度区域的测序深度,并对其进行标准化,从而得到CNV 区域的信息。基于Control-FREEC的分析结果,分别对CNV偏高(gain)和偏低(loss)的区域数量和长度进行统计。

表9 拷贝数变异(CNV)数量统计

表10 拷贝数变异(CNV)类型统计
4 个亲本拷贝数偏低的区域均超200 个,偏高的区域在79~100 个之间,所有品种在基因上游、外显子、UTR5、基因间区均有拷贝数变异,其中以外显子区域最多,在可变剪切位点、基因下游、上游/下游、UTR3区域未见有拷贝数变异。
3.8 所有变异类型的Circos图
使用Circos 软件将每个品种在染色体上的各种变异位点信息作到一张图上,SNP 或者Indel 使用每1 000 000 bp频率作Circos图(图4)。

图4 所有变异类型的Circos图
4 小结
通过对桂蚕8 号的4 个杂交亲本“锦”(9MN)“绣”(芙H)“壮”(7MH)“丽”(87H)开展全基因组重测序研究,获得4 个全基因组单核苷酸多态性(SNP)、插入缺失(InDel)、结构性变异(SV)和拷贝数变异(CNV),4个杂交亲本SNP总数均超过600万个位点,插入缺失(Indel)总数约140 万;在结构性变异(SV)方面,插入(INS)、缺失(DEL)、倒位(INV)、染色体内易位(ITX)和染色体间易位(CTX)类型均有存在,主要分布在基因间区、内含子、外显子、基因上游、基因下游,在可变剪切位点、UTR5、UTR3及位于一个基因的上游同时也是另一个基因的下游区间亦有分布;拷贝数偏低的区域均超200 个,偏高的区域在79~100 个之间,所有品种在基因上游、外显子、UTR5、基因间区均有拷贝数变异,其中以外显子区域最多,在可变剪切位点、基因下游、上游/下游、UTR3区域未见拷贝数变异。
