铁死亡在口腔癌治疗中的研究进展
2023-01-19田绣云张配黄庆洋周美云罗彬陈鑫如徐锦程
田绣云, 张配, 黄庆洋, 周美云, 罗彬, 陈鑫如, 徐锦程
蚌埠医学院第一附属医院口腔科,安徽 蚌埠(233000)
Dixon 在2012 年首次提出了铁死亡(ferroptosis)的概念,是细胞发生铁依赖的脂质过氧化(lipid peroxidation,LPO),是一种程序性死亡。铁死亡不具有典型的细胞坏死或凋亡的形态,表现为线粒体嵴的减少或消失、线粒体膜浓缩、密度增高,细胞膜和细胞核大小基本正常[1]。口腔癌是一种极为常见的恶性肿瘤,具有高复发率、高耐药性等特点,诱导其发生铁死亡是一种潜在的新治疗策略。本文系统介绍了铁死亡发生发展的机制以及铁死亡在口腔癌治疗方面的最新进展和研究前景,旨为口腔癌的治疗提供新的思路和方法。
1 铁死亡发生的机制
铁死亡发生机制包括谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)失活、铁离子含量增多、含磷脂的多不饱和脂肪(polyunsaturated fatty acid,PUFA)发生过氧化等(图1)。细胞内脂质过氧化产物如活性氧(reactive oxygen species,ROS)积累、4-羟基壬烯醛及丙二醛(malondialdehyde,MDA)含量显著增加[2]。脂质过氧化的毒性产物及ROS 的含量过高会造成DNA 和RNA 的变性降解、脂质的损伤及蛋白质活性下调。当下已研制出的铁死亡诱导剂包括柳氮磺胺吡啶、Erastin、Ras 选择性致死小分子3(Ras-selective le-thal small molecule 3,RSL3)等,铁死亡抑制剂如铁抑素-1、Liproxstatin-1 和维生素E 等物质是通过抑制脂质ROS、MDA等过氧化物的产生从而抑制铁死亡。
1.1 谷胱甘肽及氨基酸代谢异常
糖、脂肪和氨基酸等营养物质不能直接扩散进入到细胞内,它们必须通过特定转运体才能穿过细胞膜。细胞膜上存在氨基酸跨膜转运系统,如胱氨酸/谷氨酸逆向转运系统(system Xc-)由溶质载体家族SLC7A11 和SLC3A2 构成,可排出或摄取谷氨酸(glutamate)和胱氨酸(cystine)并维持两者动态平衡的系统,胱氨酸在细胞内被还原成半胱氨酸(cysteine,Cys),参与谷胱甘肽(glutathione,GSH)的合成[3]。GPX4 将GSH 转化为氧化型谷胱甘肽(oxidized glutathione,GSSG),过氧键转化为羟基,毒性产物L-OOH 还原为相应L-OH。
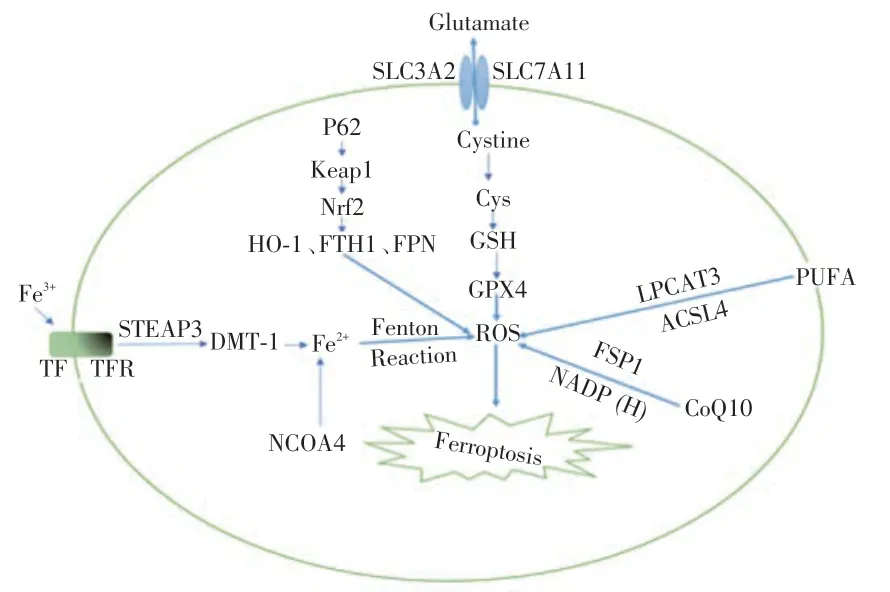
Figure 1 Mechanisms associated with ferroptosis图1 铁死亡相关机制
铁死亡诱导剂Erastin 主要抑制system Xc-中的SLC7A11 的功能;铁死亡诱导剂RSL3 直接或者间接抑制GPX4 的活性,脂质ROS 积累,最终细胞发生铁死亡[4]。Dixon 等[5]研究表明Necrostatin-1 等坏死抑制剂和氯喹或3-甲基腺嘌呤等自噬抑制剂都不能抑制Erastin 和RSL3 诱导的细胞死亡。Sun等[6]表示Fin56 是目前最新的铁死亡诱导剂,与激活细胞发生自噬联合应用可发挥协同效应,主要促进GPX4 的降解,然而这种降解的机制目前还不完全清楚。
1.2 铁离子代谢异常
为了满足新陈代谢需求和促进自身生长,肿瘤细胞对铁的需求比正常的细胞更大,这使它们更容易受到铁死亡的影响[7]。细胞中Fe2+是携带Fe3+的转铁蛋白(transferrin,TF)与细胞膜上的转铁蛋白受体(transferrin receptor,TFR)相结合,经6 次跨膜前列腺跨膜上皮抗原3(six-transmembrane epithelial antigen of the prostate 3,STEAP3)还 原 为Fe2+,再由二价金属转运蛋白-1(recombinant divalent metal transporter-1,DMT-1)运输到细胞质内;由铁蛋白被核受体共激活因子4(nuclear receptor coactivator 4,NCOA4)经自噬溶酶体降解后释放出Fe2+。细胞内的Fe2+部分存在于线粒体不稳定铁池中,部分储存在铁蛋白重链(ferritin heavy chain 1,FTH1)和铁蛋白轻链组成的铁储蛋白复合物中。当细胞发生铁超载时,过多的游离Fe2+与过氧化氢(H2O2)发生芬顿反应产生大量毒性产物和ROS,促使细胞发生铁死亡[8]。
Keap1 是一种底物衔接蛋白,在细胞质内主要和核因子红细胞相关因子2(NF-E2-related factor 2,Nrf2)相结合,正常情况下在细胞质中通过泛素化与Nrf2 相结合。p62 在Nrf2 处相互竞争Keap1 上的结合位点,当细胞处于氧化应激时,P62 与Keap1结合,Nrf2 与Keap1 泛素化断裂,Nrf2 则由胞质转移至细胞核中,从而使细胞质内的Nrf2 降低[9]。Nrf2 的下游靶基因包括膜铁转运蛋白(ferroportin,FPN)、FTH1、血红素加氧酶-1(heme oxygenase-1,HO-1)和醌氧化还原酶1(NAD(P)H quinine oxidoreductase 1,NQO1)等的下调,会使细胞内游离铁离子增多,抗氧化应激能力降低,ROS 增多[10-11]。ROS 的过量生产也会攻击铁蛋白和含铁的蛋白质,导致不稳定的铁释放。因此Nrf2 可作为抗氧化调节剂可用来阻止铁死亡的发生。Li 等[12]的研究已表明上调Nrf2/HO-1 通路可减轻细胞发生铁死亡。
1.3 脂质代谢异常
游离的PUFA 可被酰辅酶A 合成酶长链家族成员4(Acyl-CoA synthetase long-chain family member 4,ACSL4)和溶血磷脂酰胆碱酰基转移酶3(lysophosphatidylcholine acyltransferase 3,LPCAT3)激活并入磷脂膜中发生脂质过氧化[13]。研究表明在PUFA 相关磷脂中含有花生四烯酸(arachidonic acid,AA)以及肾上腺素基团磷脂酰乙醇胺(phosphatidyl ethanolamines,PEs)的部分被证明是细胞发生脂质过氧化的重要部分[14]。
1.4 其他影响铁死亡的通路
铁死亡抑制蛋白1(ferroptosis suppressor protein 1,FSP1)作为氧化还原酶经豆蔻酰化被募集到质膜(细胞膜、内质网、高尔基体等)上,FSP1 还原辅酶Q10(coenzyme Q10,CoQ10)阻止脂质过氧化产物的致死性积累。研究表明胱氨酸也可增加细胞内CoQ10 的含量。FSP1/CoQ10/NAD(P)H 通路作为一个平行系统存在,即能够独立于GPX4 通路抑制铁死亡,又可与GSH-GPX4 协同工作抑制磷脂过氧化和铁死亡[15]。
2 铁死亡与口腔癌的治疗
口腔癌在全球每年新增约60 万例,死亡率为40%~50%。人类乳头瘤状病毒(human papillomavirus,HPV)、咀嚼槟榔、吸烟等均为口腔癌发生的高危因素,常伴有严重疼痛、出血破溃、咬合关系错乱、口腔异味明显,晚期可出现面部畸形、组织坏死、颌骨骨折等症状,易发生远处转移,局部复发率高,患者预后较差,生活质量较低[16-17]。虽然使用了手术、药物以及放化疗等各种方法联合治疗口腔癌,但癌细胞仍易产生较高的放化疗耐药性,5 年生存率仍较低。
目前铁死亡作为一种肿瘤抑制机制,被视为人类疾病的潜在治疗方法。细胞抗氧化功能与癌症进展相关,口腔癌细胞发生铁死亡导致其抗氧化应激能力降低,即细胞内产生氧气的氧化剂和抗氧化剂之间平衡失调,造成线粒体以及内质网、高尔基体、溶酶体等功能障碍,细胞内相关的基因组不稳定,GPX4、HO-1、Nrf2、SLC7A11 等抗氧化剂被抑制,产生大量毒性物质和ROS,过量的ROS 使癌细胞持续处于氧化应激的状态,最终发生癌细胞死亡以及实体肿瘤逐渐消退。这种细胞死亡机制不需要caspase 激活,也不需要其他凋亡效应因子(如Bax 或BAK)的参与。外源性添加多不饱和脂肪、葡萄糖或胰蛋白酶等可增加细胞对铁死亡的敏感性。已有研究表明铁死亡与诸多癌症相关,如肺癌、膀胱癌、肝癌等[4,6,9],激活癌细胞发生铁死亡可有效抑制肿瘤增殖,增强放疗、化疗以及免疫治疗的效果。
2.1 铁死亡与耐药性
化疗药物(如顺铂、卡铂等)的高耐药性是肿瘤复发和疾病特异性死亡的重要驱动因素。具有化疗耐药性的癌细胞获得间充质特征便允许发生侵袭和迁移,导致癌症患者的临床治疗效果不佳。癌细胞暴露于化疗药物中,高浓度ROS 的产生抑制癌细胞的增殖,而癌细胞启动某种机制抵抗ROS 的产生因此产生耐药。研究表明顺铂耐药性与癌细胞氧化应激后的代谢恢复有关[18],如糖代谢、脂肪酸代谢、氨基酸代谢。最近一些学者观察到了顺铂可作为铁死亡的诱导剂,与Erastin 联合治疗恶性肿瘤存在显著的协同效应,一些易转移的间充质癌对化疗药物有高度耐药性,但对铁死亡敏感性高[19]。Lee 等[20]表明谷氧还蛋白5(glutaredoxin 5,GLRX5)沉默后细胞内游离铁离子增加,ROS 积累,耐药口腔癌细胞发生铁死亡。Roh等[21]表明青蒿琥酯可通过抑制Nrf2/ARE 通路,导致细胞内GSH 耗竭以及ROS 积累而发生铁死亡,并逆转耐药癌细胞的铁死亡抗性。部分siRNA 和shRNA 也通过抑制癌细胞中的system Xc-致GSH耗竭,从而导致顺铂耐药癌细胞发生铁死亡。You等[19]表明抑制线粒体丙酮酸载体1(mitochondrial pyruvate carrier 1,MPC1)的缺失可下调GPX4 或抑制xCT 杀死耐药性口腔癌细胞,同时破坏抗氧化系统,增加了线粒体ROS 的产生和脂质过氧化发生。Sato 等[22]表明非热等离子体(non-thermal plasma,NTP)可特异性使口腔癌细胞发生铁死亡,在放射治疗和化疗完成之前应用NTP 作为一种附加治疗是可行的。Sun 等[23]表明癌细胞中YES 关联蛋白(Yes-associated protein,YAP)/具有PDZ 结合基序的转录辅激活子(transcriptional coactivator with PDZ binding motif,TAZ)为铁死亡相关通路之一,但其过度激活或功能突变会提高癌细胞对治疗药物产生耐药性。
2.2 铁死亡与免疫功能
口腔癌也属于免疫抑制性疾病,癌细胞通过免疫抑制细胞因子积聚、细胞活性和抗原呈递功能受损、T 细胞耗竭等逃避免疫监视和抗肿瘤免疫反应。诱导癌细胞发生铁死亡可激活免疫细胞的能力,可作为增强免疫治疗活性的策略。巨噬细胞是肿瘤微环境(tumor microenvironment,TME)中的主要吞噬细胞和抗原呈递细胞,M1-M2 巨噬细胞极化系统中,M1 亚型在TME 中有高水平的肿瘤坏死因子、组织相容性复合体2(major histocompatibility complex2,MHC2)或诱导型一氧化氮合酶的情况下被激活,M2 亚型在TME 中存在高水平的精氨酸酶1(arginase 1,ARG1)、IL-10 等的情况下被激活。肿瘤相关巨噬细胞(tumor-associated macrophages,TAM)为M2 型,将TAM 从M2 表型复极化为M1 表型可提高免疫治疗的疗效[24]。SOCS1 可下调p53 靶基因和SLC7A11 的表达诱导铁死亡,在口腔癌中FTHI 的表达与M2 型巨噬细胞的浸润呈现为正相关,增加SOCS1 的表达或降低FTH1 的表达可诱导口腔癌细胞发生铁死亡;铁死亡相关因子SOCS1 与M1 巨噬细胞存在某种关联,但其具体的调控机制尚未明确,研究者推测其可能通过SOCS1和FTH1 保持动态平衡共同调控肿瘤的增殖[25]。癌细胞释放的细胞因子高迁移率族蛋白B1(highmobility group protein 1,HMGB1)可以促进巨噬细胞向M1 型极化,HMGB1 缺失可抑制铁死亡的发生。部分免疫系统可能通过铁死亡介导CD8+T 细胞分泌IFN-γ 抑制肿瘤活性,同时与免疫检查点阻断发挥协同作用,CD8+T 细胞可通过调控IFN-γ 的产生来抑制system Xc-中的SLC7A11 和SLC3A2 亚基诱导癌细胞发生铁死亡。IFN-γ 在特定情况下可上调细胞程序性死亡配体1(programmed cell death-ligand 1,PD-L1)的表达帮助癌细胞进行免疫逃逸,用PD-L1 阻断剂和GSH 缺失治疗可激活T 细胞的抗肿瘤活性,并在体内诱导癌细胞铁死亡[26]。Li 等[27]表明口腔癌患者因免疫B 细胞抑制肿瘤生长以及树突状细胞数量显著增高而激活的免疫反应都与铁死亡有关。
2.3 铁死亡与治疗药物
姜黄素类似物、奎尼司坦、青蒿素衍生物等药物可诱导口腔肿瘤发生铁死亡。吉非替尼和二甲双胍可能是治疗复发或晚期口腔癌潜在新药[28]。研究表明组蛋白去乙酰化酶抑制剂通过抑制GPX4 相关通路而诱导口腔鳞状细胞癌(oral squamous cell carcinoma,OSCC)中CAL-27、TCA-8113 细胞发生铁死亡[29]。Cai 等[30]表明雷公藤甲素(triptolide,TPL)抑制线粒体己糖激酶Ⅱ(hexokinase-Ⅱ,HK-Ⅱ)和有氧糖酵解促进口腔癌细胞发生铁死亡,TPL 与Erastin 的联合使用在体外和裸鼠模型中对抑制肿瘤存活方面发挥了强大的协同效应。与传统药物相比,配制纳米药物载体可以克服化疗药物的溶解度问题和有限的膜通透性,并且具有较好的生物相容性。Qian 等[31]表明纳米材料可以通过参与生化反应和干扰代谢平衡来最大限度地促进活性氧的形成,诱导细胞发生铁死亡,另一方面,它们可特异性直接攻击肿瘤组织,是目前在癌症治疗领域的研究热点,但其临床转化方面仍不尽如人意。
2.4 铁死亡与治疗靶点
p53 已被证明是细胞生长、增殖和铁死亡的中心调节因子,诱导受损细胞凋亡来保护细胞免受致癌转化,p53 对凋亡的调控涉及细胞对线粒体呼吸作用和脂质过氧化变化的反应。Fukuda 等[32]表明当p53 基因点突变功能丧失后,GPX4 会激活,铁死亡被抑制,因此促进了口腔肿瘤的生长。GPX4作为哺乳动物中最重要的抗氧化和脂质修复酶之一,当细胞内SLC7A11 较低时,GPX4 因底物GSH合成不足而失活,细胞发生脂质过氧化。肿瘤抑制因子p53 和BAP1 下调SLC7A11 基因以及MRP1在癌细胞中的高表达均可抑制GSH 的摄取从而促进癌细胞株发生铁死亡[33]。SLC7A11 可作为一个治疗口腔癌的潜在的靶向标志物。Hémon 等[34]表明在HPV 阳性的口腔癌患者中,SLC7A11 低表达比SLC7A11 高表达对铁死亡更敏感。卵巢肿瘤家族成员泛素醛结合1(ovarian tumor family member deubiquitinase,ubiquitin aldehyde binding 1,OTUB1)被确定为SLC7A11 稳定因子,OTUB1 失活可使肿瘤细胞对铁死亡敏感[33],但在口腔癌治疗中的应用需进一步研究。环状RNA(circular RNA,CircRNA)对OSCC 发生发展的调控作用,通过microRNA 调控内源性RNA,或通过调节基质金属蛋白酶9 相关mRNA 的稳定性促进OSCC 的转移[35-36]。CircRNA_100290 靶向miR-378a,通过葡萄糖转运蛋白1 和糖酵解调节OSCC 进展[37]。Yang 等[38]表明circFNDC3B调节miR-520d-5p靶向增加SLC7A11 以抑制OSCC 细胞铁死亡,从而促进肿瘤增殖,SLC7A11与miR-520 家族其他成员是否具有相关性仍需要进一步研究,如miR-520c-5p、miR-520f-3p、miR-520a-5p 等。
Fox 等[39]表明Nrf2 激活促进休眠肿瘤细胞的复发,与不良预后相关。Hu 等[40]表明Nrf2 下游靶点FTH1 在口腔癌中的表达比正常组织中高,颈部淋巴结转移和预后不良与FTH1 的上调显著相关。研究表明FTH1 作为口腔癌的独立预后因素,与M1、M2 巨噬细胞浸润相关[25]。FTH1 表达上调与大多数实体瘤中的巨噬细胞浸润呈正相关[41],其与预后的相关性及是否可作为口腔肿瘤治疗的靶点仍需进一步研究。
本综述系统介绍了铁死亡的机制及其在口腔癌治疗方面的应用,希望有助于科研人员进行更深一层的研究,如研发新药和新治疗机制,最终为临床治疗口腔癌做出贡献。
【Author contributions】 Tian XY collected the references and wrote the article. Zhang P, Huang QY, Zhou MY, Luo B, Chen XR revised the article. Xu JC guided the writing of the article. All authors read and approved the final manuscript as submitted.
