原子吸收光谱测定味精中锌元素不同前处理方法的比较
2023-01-13陈文彬
陈文彬
上海工微所科技有限公司,上海 200233
锌是人体必需的微量元素之一,在人体生长发育、生殖遗传、免疫、内分泌等重要生理过程中起着极其重要的作用[1-2]。人体缺锌会引起许多疾病, 摄入过量的锌亦有不利的影响。摄入含有过量锌的食物和饮料会引起锌中毒[3]。大量的锌还能抑制吞噬细胞的活性和杀菌力,从而降低人体的免疫功能,使抵抗力减弱,对疾病易感性增加[4]。过量补锌可降低机体内血液、肾和肝内的铁含量,从而使人体发生顽固性缺铁性贫血[5]。我国味精的主要制作工艺是将谷物淀粉水解为葡萄糖配合菌种发酵大量积累谷氨酸,再通过锌盐法提取发酵液中的谷氨酸,最终的谷氨酸中和液会经过硫化钠法或树脂法进行除锌处理。为了验证产品的除锌效果,防止味精中的锌对食用者造成危害,对成品味精进行锌含量的测定是非常必要的。然而,目前现行的味精标准GB 2720—2015中并没有规定味精中锌的检测方法。国内见于报道的味精中锌的检测方法有原子吸收光谱法、二硫腙比色法[6]等。
本实验根据GB/T5009.14—2017食品中锌的测定第一法原子吸收光谱法,并对该方法的前处理进行了改进,使其更加适合味精样品的测定。首先,硝酸-高氯酸的湿法消解方法虽然得到广泛的运用,但是高氯酸本身存在可能爆炸的潜在危险,业内也出现过使用高氯酸发生爆炸的事故。此外,高温干法灰化虽然对于破坏样品中有机质是很好的手段,并且能降低酸污染,但灰化温度过高易造成金属元素的挥发或吸附于坩埚上;温度太低又会因为灰化不彻底而影响测定结果[7]。因此,本文尝试改变常规干法消解使用的酸和加入酸的时机、以及尝试使用不同的酸进行湿法消解和微波消解,对样品消解时间、消解效果和实验结果的精密度和回收率的影响进行分析,最终得出较可靠和高效率的消化方法。
1 材料与方法
1.1 试验材料与试剂
味精:1号品牌99%细晶味精,1号品牌80%粉状味精,2号品牌99%味精。
标准溶液和试剂:水中锌0.100 g/L标准品(上海市计量测试技术研究院),盐酸,硝酸,高氯酸,实验所用试剂均为优级纯,实验用水为去离子水。
1.2 试验仪器与设备
原子吸收分光光度计(Agilent Technologies 200 Series AA),ECH-II微机控温加热板(上海新仪微波化学科技有限公司),MDS-6微波消解仪(上海新仪微波化学科技有限公司),立式万用电炉(上海慧泰仪器制造有限公司),SX2-410马弗炉(上海实研电炉有限公司)。
1.3 试验方法
消解用酸的选择,事先拟定三种不同的消解用酸,混合酸:硝酸+高氯酸(4+1);硝酸 ;盐酸。
1.3.1试样前处理
(一) 先灰化再加酸的干法消解
准确称取0.5 g样品置于50 mL瓷坩埚中,小火炭化至无烟后移入马弗炉中,500 ℃±25 ℃灰化约8 h后,取出坩埚,再加入5 mL硝酸或盐酸,小火加热,不使干涸,如此反复处理,直至残渣中无炭粒,待坩埚稍冷,用盐酸(1+11)溶解洗涤移入25 mL容量瓶中[8],定容。并且按同一操作方法做试剂空白试验。
(二) 先加酸再灰化的干法消解
准确称取0.5 g样品置于50 mL瓷坩埚中,加入5 mL消解用酸,放置0.5 h,小火蒸干,继续加热炭化,移入马弗炉500 ℃±25 ℃灰化约1 h后,取出,加1 mL消解用酸,小火蒸干,再移入马弗炉500 ℃±25 ℃灰化约1 h,取出,用盐酸(1+11)溶解洗涤移入25 mL容量瓶中,定容。并且按同一操作方法做试剂空白试验。
(三) 湿法消解
准确称取1 g样品于高脚烧杯中,加入10 mL消解用酸加盖浸泡过夜,在电热板上加热消解至冒白烟,消化液呈无色透明或略带黄色,放冷,用去离子水转移洗涤至25 mL容量瓶,定容。并且按同一操作方法做试剂空白试验。
(四) 微波消解
准确称取0.5 g样品置于微波消解罐中,加入5 mL消解用酸,置于电热板上120 ℃预消化45 min(防止在微波消解时反应,短时间内罐内压力过大,造成安全装置自动放气减压,使微量元素损失[9]),放冷,视溶液体积补加1 mL~2 mL消解用酸,放入微波消解仪中逐步加压 消解(消解仪设定参数见表1),消解完全后,取出,放电热板上赶酸,放冷,用去离子水转移洗涤至25 mL容量瓶,定容。并且按同一操作方法做试剂空白试验。
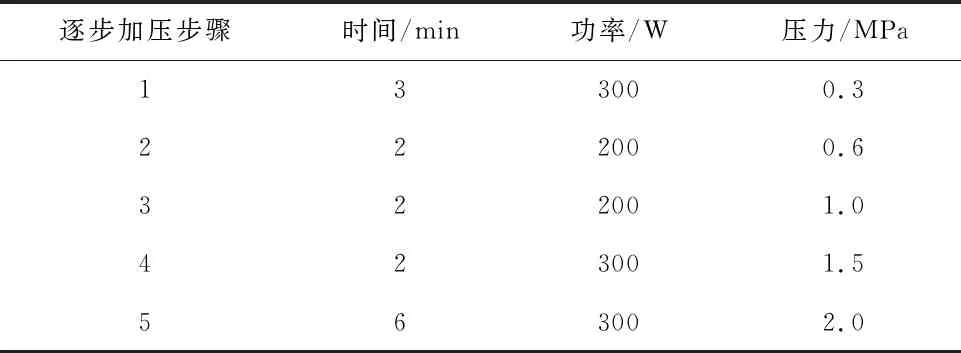
表1 微波消解仪设定参数
1.3.2样品测定
准确吸取0 mL,0.50 mL,1.00 mL,2.00 mL,3.00 mL和4.00 mL锌标准使用液(5 μg/mL),分别置于25 mL容量瓶中,以1 mol/L盐酸稀释至刻度,混匀(各容量瓶中每毫升分别相当于0 μg,0.1 μg,0.2 μg,0.4 μg,0.6 μg,0.8 μg锌)。
将处理后的样液、试剂空白液和各容量瓶中的锌标准溶液分别导入调至最佳条件的火焰原子化器(火焰原子吸收分光光度计测定工作条件见表2),先测定标准系列的吸光度,得到锌元素标准系列回归方程,再将样品测定吸光度代入回归方程,计算出味精样品中锌元素的含量。

表2 火焰原子吸收分光光度计测定工作条件
2 结果与讨论
对三种不同味精进行上述方法的测定,发现三种味精中锌含量均小于定量限。因此对几种消解方法做如下实验:(1)对空白样品吸光度进行20次平行测定,计算方法检出限。(2)在三种不同的味精样品中分别加入0.50 mL、1.00 mL、2.00 mL锌使用液(5 μg /mL),分别为2.5 μg、5.0 μg、10 μg锌进行加标实验,每个浓度做6个平行。(3)对三种味精加入2.5 μg锌的样品重复测定6次,验证精密度。
(一) 先灰化再加酸的干法消解
前文提到高氯酸具有加热易爆的特性,而干法消解的加热温度较高,所以本文试验了用拟定的硝酸和盐酸两种酸来进行实验,测定该方法的检出限,精密度,和加标回收率。结果见表3和表4。

表3 先灰化再加硝酸的干法消解(n=6)
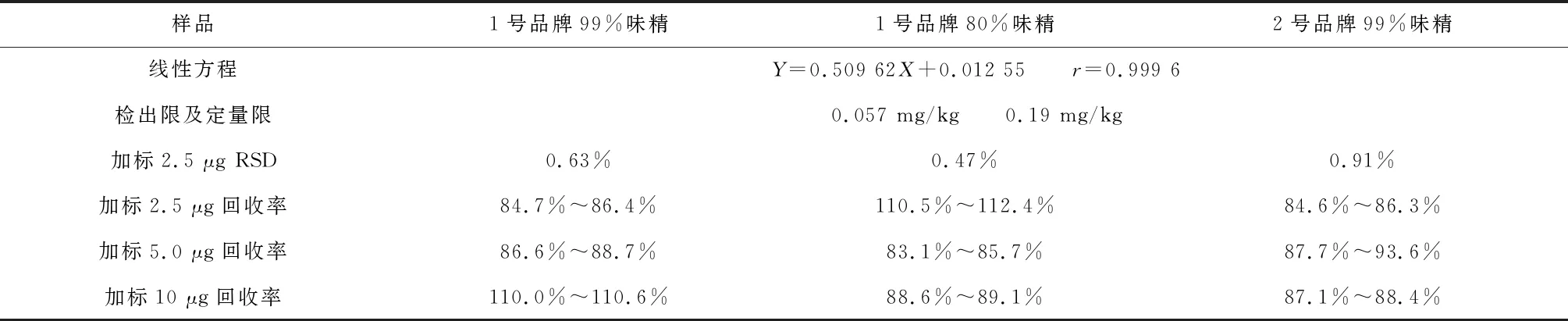
表4 先灰化再加盐酸的干法消解(n=6)
从表中数据可以看出干法消解的精密度较好,但是由于高温加热和灰化时间过长,容易导致锌的损失,回收率勉强满足分析要求但是不稳定,高温加热味精还会生成有轻微毒性的焦谷氨酸钠。因此,干法消解可以用硝酸和盐酸来代替高氯酸,但过程繁琐,消耗时间长,效率低。
(二) 先加酸再灰化的干法消解
由于干法消解耗时很长,所以本文试验了先在1号品牌99%味精中加入拟定的三种酸再灰化来加快反应速率,并测定该方法的检出限,精密度,和加标回收率。干法消解完全后样品如图1,测定结果见表5。

图1 从左到右依次为硝酸+高氯酸(4+1),硝酸,盐酸
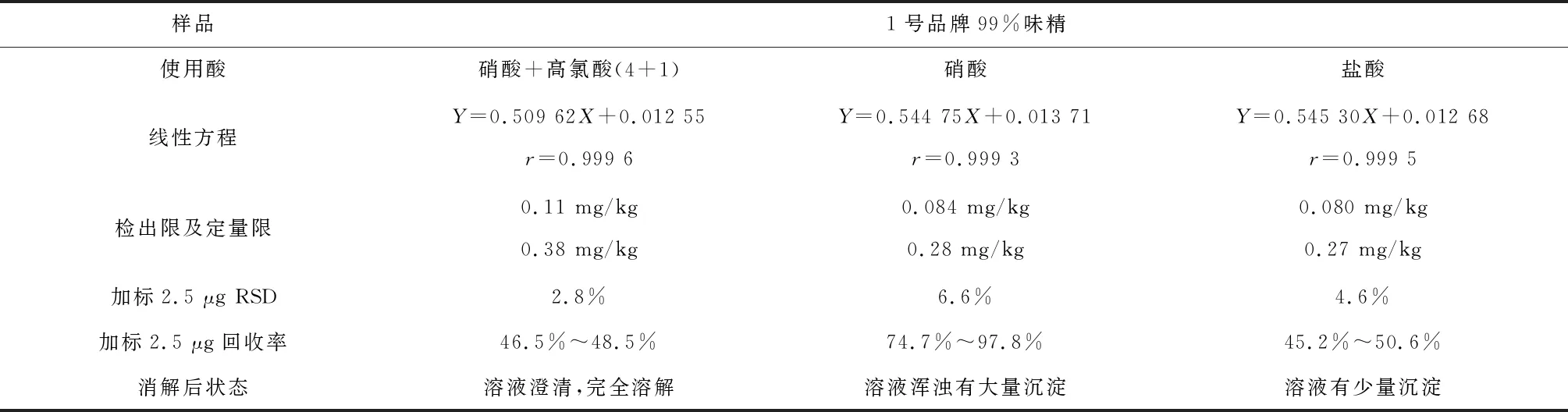
表5 1号品牌99%味精先加酸再灰化的干法消解(n=6)
从图1和表5数据中可以看出,以上三种酸可以将样品消解完全,其中混合酸酸消解效果最佳,但是味精加入酸后小火加热反应剧烈,从结果来看混合酸和盐酸回收率很低,说明样品中锌在剧烈反应中大量损失,硝酸回收率偏差大,说明用硝酸消解的稳定性不好。此外,虽然该方法在电炉炭化过程比前法节省了时间,但是三种酸的灰化过程都要进2次马弗炉,由于马弗炉升温和冷却时间较长,导致该方法总体消耗的时间与前法相比没有显著减少,实验效率也没有提升。证明该方法不适用于味精的锌的消解。
(三) 湿法消解
姚正堂等[10]的研究证实了硝酸+高氯酸(4+1)湿法消解也能运用于味精中锌的测定,本文尝试用拟定的硝酸和盐酸来代替硝酸+高氯酸的混合酸,经试验取样5 g消解完全后烧杯底部溶液浓度过高,液体粘稠,不利于转移洗涤,加标回收率为74.0%~84.3%。因此取样1 g进行试验,并测定该方法的检出限,精密度,和加标回收率,是否能达到更好的效果。结果见表6和表7。

表6 硝酸湿法消解(n=6)

表7 盐酸湿法消解(n=6)
从实验数据可以看出使用硝酸和盐酸进行湿法消解实验结果无明显差异,两种酸都能将味精消解完全,两种方法的精密度和准确度也都达到分析要求,说明在不使用高氯酸的情况下,硝酸和盐酸本身也能胜任味精的消解,且整个实验耗时较少,效率较高,安全可靠。
(四) 微波消解
微渡消解技术是一种崭新的、高效的样品消解方法[11]。本文分别用拟定的混合酸,硝酸和盐酸来进行消解,并测定该方法的检出限,精密度,和加标回收率,比较三种酸对结果的影响。测定结果见表8、表9和表10。

表8 混合酸微波消解(n=6)

表9 硝酸微波消解(n=6)

表10 盐酸微波消解(n=6)
从实验数据中可以看出三种酸均能将味精消解完全,三种酸的实验结果无明显差别,同时也拥有较好的精密度和加标回收率,其中唯一缺点是混合酸的样品在经过微波后罐体温度极高,放气也很猛烈,具有一定危险性。其余两种酸的消解都比较稳定。试剂用量少,减少酸的污染,操作简单。
综上所述,从图2中7种消解方法的比较可以看出干法消解回收率不稳定,且过程繁琐,消耗时间长,效率低,不能满足味精中锌的测定要求。对姚正堂等[10]湿法消解进行了改进,分别使用了硝酸和盐酸来代替混合酸,其中使用硝酸相对标准偏差在1.3%~3.1%,回收率在94.8%~110.1%,盐酸相对标准偏差在0.9%~3.8%,回收率在98.2%~106.7%结果准确可靠。尝试了使用三种不同的酸进行微波消解,其中混合酸相对标准偏差0.44%~0.67%,回收率在91.4%~106.2%,硝酸相对标准偏差0.6%~2.6%,回收率在96.8%~111.7%,盐酸相对标准偏差1.3%~3.3%,回收率在90.9%~101.2%。说明微波消解效果很好,唯一缺点是混合酸微波消解有一定危险性。
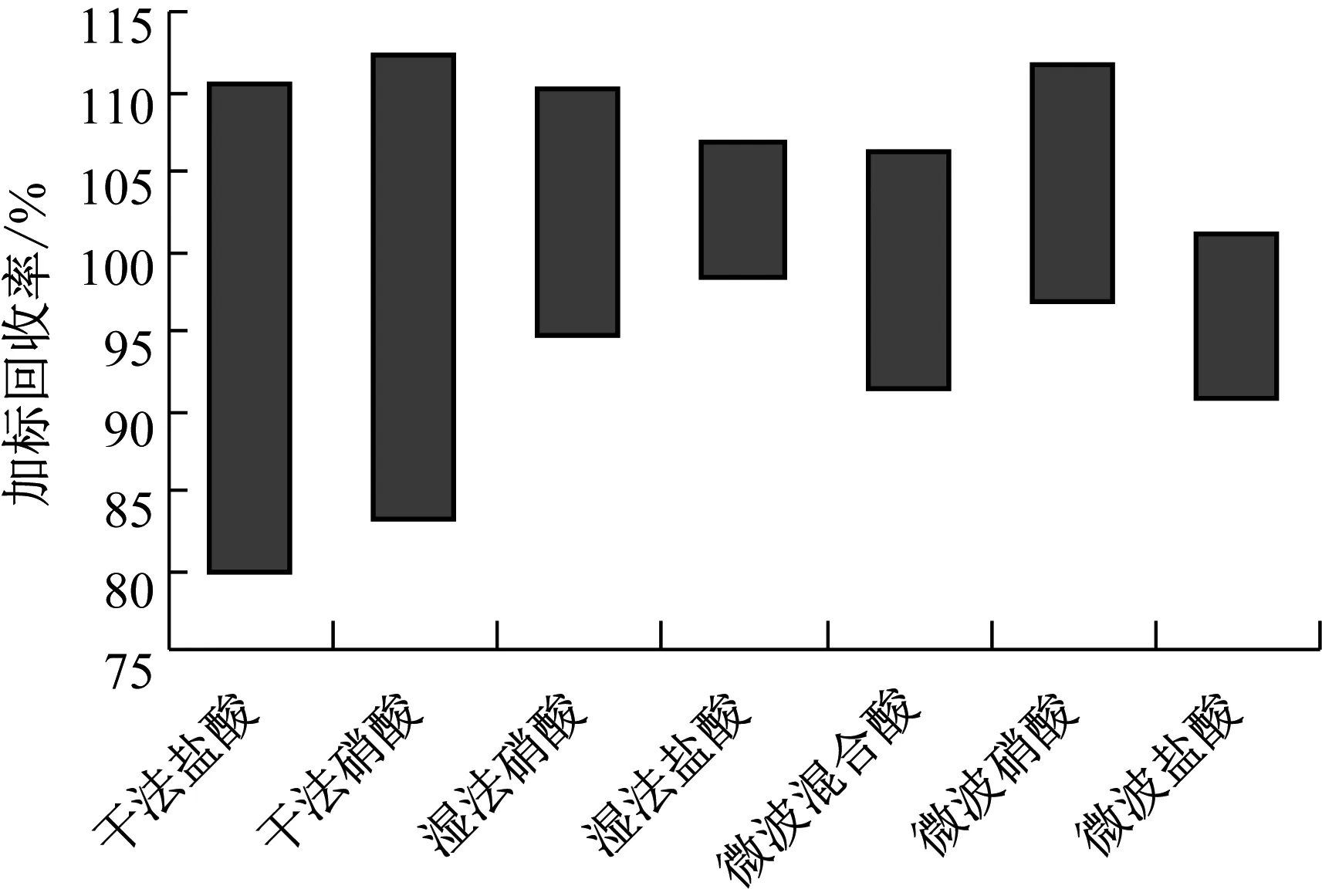
图2 7种消解方法加标回收率
3 结论
本文对味精中锌的测定方法进行了改进,使用纯硝酸和纯盐酸来代替混合酸进行干法和湿法消解,并且使用三种酸进行了微波消解。标准曲线在0.1 μg/mL~0.8 μg/mL浓度范围内线性良好,相关系数大于0.999,三种味精的锌用几种方法测定都小于定量限,三种方法中干法消解回收率不稳定,湿法和微波消解结果精密度和准确度则较好,同种方法测定不同的99%味精和80%味精回收率无明显差异,说明本文尝试的湿法和微波消解适用于不同味精中锌的测定。在三种酸的比较中,使用盐酸进行湿法和微波消解效果最佳,使用盐酸进行湿法消解的加标相对标准偏差在0.9%~3.8%,回收率在98.2%~106.7%。使用盐酸进行微波消解的加标相对标准偏差1.3%~3.3%,回收率在90.9%~101.2%。使用微波消解在少量样品时效率高,湿法消解则适用于大批量的样品。
