MSMEG_0372基因敲除、回补及过表达耻垢分枝杆菌的构建与鉴定
2023-01-10刘仪牛宏红杨赛丽曹旭东袁俐
刘仪,牛宏红,杨赛丽,曹旭东,袁俐
(石河子大学医学院病原生物学与免疫学教研室,新疆 石河子 832000)
结核病(Tuberculosis,TB)是由结核分枝杆菌(Mycobacteriumtuberculosis,M.tuberculosis)引起的严重危害人类健康的慢性传染病。根据世界卫生组织(World Health Organization, WHO)的报告,2020年全球新发TB患者987万,其中我国新发TB病例为84.2万,在30个TB高负担国家中排第2位,仅次于印度(259万)[1]。而耐药TB一直是TB临床诊断和治疗的重点和难点[2]。
M.tuberculosis细胞壁中的脂质成分可占细胞壁干重的60%以上,其中分枝菌酸与M.tuberculosis的存活、致病性、毒力、免疫逃避和生物膜形成密切相关,在抗TB研究中有着极其重要的地位[3]。M.tuberculosis的脂肪酸合成 (fatty acid synthesis, FAS) 途径有两种形式:Ⅰ 型脂肪酸合成 (FAS-Ⅰ)途径和 Ⅱ 型脂肪酸合成(FAS-Ⅱ)途径。FAS-Ⅰ是一个多功能且高效的催化酶,可以完成脂肪酸碳链延伸的所有步骤,而FAS-Ⅱ 主要由FabG1 (MabA,Rv1483), HadAB/HadBC (Rv0635-0637)、InhA (Rv1484) 和KasA/KasB (Rv2245/Rv2246)4个酶组成,他们在碳链延伸的过程中发挥着不同的催化作用,也是抗结核药物常见的靶标[3]。FabG4(Rv0242c)是FabG1的同源蛋白蛋白之一,在所有分枝杆菌中保守[4],其功能可能与FabG1相似[4]。FabG4与FabG1的辅酶分别是NADH和NADPH,而NADH相比NADPH是一种低能量分子[4]。FabG4对在罗辛甘油基本培养基(Roisin’s glycerol minimal media)上的分枝杆菌的存活至关重要[4]。FabG4在亚抑菌浓度链霉素胁迫下表达增高[4],也在M.tuberculosis的气-液界面的生物膜中特异表达[4]。所以,人们推测FabG4在营养受限或抗生素压力下会参与一些与膜代谢相关的低能耗途径[4]。同时,FabG4具有抗原特性[4],对MHC-I和MHC-II分子具有高亲和力[5],有望成为分枝杆菌生物膜的特异标记物[4]。
生物膜由细菌及其分泌的细胞外聚合物组成。生物膜形成可以导致M.tuberculosis表型耐药[6],所以深入研究FabG4的作用对于耐药TB的诊断和治疗具有重要的意义。非致病性的耻垢分枝杆菌(Mycobacteriumsmegmatis,M.smegmatis)是一种常见的能够替代M.tuberculosis的模式菌[7]。M.tuberculosisRv0242c在M.smegmatis中的直系同源基因是MSMEG_0372,二者具有78.9%的基因同源性。目前尚无有关MSMEG_0372基因功能的研究报道。为了研究该基因的功能,我们构建了MSMEG_0372基因敲除、回补及过表达M.smegmatis。
1 材料与方法
1.1 材料
1.1.1 菌株、质粒与引物
野生型M.smegmatismc2155,大肠杆菌(E.coli) DH5α和HB101,及pMV361,p0004s和phAE159质粒均购买自上海晶诺生物科技有限公司。引物见表1,表中加粗、斜体并下划线的序列为限制性内切酶Van91I的酶切位点。

表1 引物
1.1.2 主要试剂
胰蛋白胨、酵母提取物和琼脂粉购自北京索莱宝科技有限公司,氯化钠和甘油购自上海生工生物工程股份有限公司,Middlebrook 7H9、Middlebrook 7H10和ADC购自美国BD公司,潮霉素B和吐温80购自美国Sigma公司,高纯度低电渗琼脂糖购自北京擎科生物科技有限公司,Trans2K Plus II DNA Marker和Trans15K DNA Marker购自北京全式金生物技术有限公司,Fast Digest Van91I、限制性内切酶PacI、Fast DigestHind III、Fast DigestEcoRI、T4 DNA连接酶和Phusion High Fidelity DNA Polymerase购自美国赛默飞世尔科技公司,氨苄青霉素钠和氯霉素购自美国Amresco,硫酸卡那霉素购自青岛MDBIO,包装试剂盒购自美国EPICENTRE Biotechnologies,质粒提取试剂盒购自北京天根生化科技有限公司,胶回收试剂盒和细菌基因组提取试剂盒购自美国Omega公司。
1.1.3 培养基
按说明书配置7H9液体培养基、7H10平板、LB液体和固体培养基。其中,7H9液体培养基、7H10平板用于M.smegmatis的培养,LB液体和固体培养基用于E.coli的培养。顶层琼脂(Top Agar):Middlebrook 7H9 粉末0.47 g、琼脂粉0.75 g,加双蒸水补充至100 mL,121 ℃高温灭菌15 min。MP缓冲液:50 mmol·L-1Tris-HCl、150 mmol·L-1NaCl、10 mmol·L-1MgSO4、2 mmol·L-1CaCl2,用稀HCl调至pH 7.5或7.8,用0.22 μm滤菌器除菌,保存备用。
1.1.4 主要仪器
生物安全柜(BSC-1304ⅡA2)购于苏州安泰空气技术有限公司,恒温培养箱(GSP-70)购于天津市莱玻特瑞仪器设备有限公司,凝胶成像仪(WD-9413B)和电泳仪(DYY-6C)购于北京六一生物科技有限公司,电子天平(PL1502E)和酸度计(FE28)购于瑞士梅特勒-托利多公司,PCR 仪(S1000)和电穿孔仪(Gene Pulser MXcell)购于美国伯乐公司。
1.2 方法
1.2.1 构建p0004s-AES质粒及鉴定
根据MSMEG_0372基因及其上下游的DNA序列,设计该基因的左臂引物(LFP/LRP)和右臂引物(RFP/RRP),左、右臂是等位基因同源重组的底物(allele exchange substrates,AES)(图1)。以M.smegmatis的基因组DNA为模板(2 μL),分别以LFP/LRP引物对和RFP/RRP引物对为引物(F和R各2.5 μL),加入高保真聚合酶Phusion High Fidelity DNA Polymerase(0.5 μL),5×Phusion HF Buffer(10 μL),DMSO(1.5 μL),10 mmol·L-1dNTP(1 μL),补充无核酸酶水制备50 μL反应体系。PCR条件:98 ℃ 30 s;98 ℃ 10 s,58 ℃ 30 s,72 ℃ 30 s,共30个循环;72 ℃ 10 min。通过琼脂糖凝胶电泳对PCR扩增产物进行鉴定。用限制性内切酶 Van91I酶切MSMEG_0372基因的左、右臂(AES)并用胶回收试剂盒回收酶切产物。用天根质粒提取试剂盒提取p0004s质粒,用限制性内切酶 Van91I对其酶切并回收其中的两个大片段(约3.6k bp和1.6k bp)产物。将左、右臂(AES)及p0004s质粒的两个大片段用T4 DNA 连接酶连接起来,构建p0004s-AES质粒,转化其到E.coliDH5α感受态细胞中,在含75 μg·mL-1潮霉素B的LB平板上筛选阳性菌落。挑取阳性菌落,摇菌,收集菌体,提取p0004s-AES质粒并通过DNA测序鉴定。
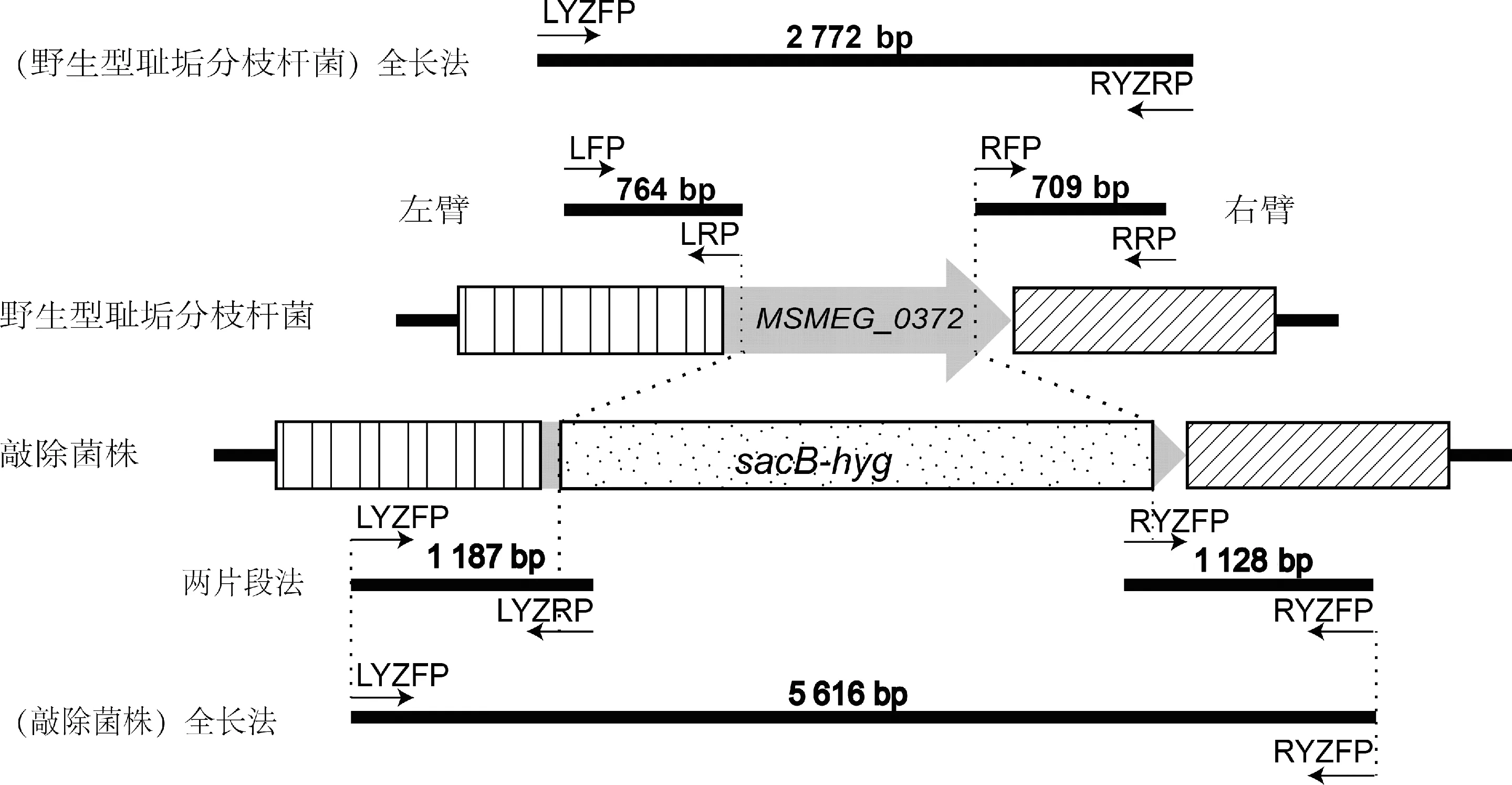
图1 MSMEG_0372基因敲除菌株引物示意图
1.2.2 构建phAE159-AES噬菌粒
用Omega 质粒提取试剂盒提取phAE159质粒以及p0004s-AES质粒,分别用PacI酶切并回收产物。将回收片段用T4 DNA 连接酶连接起来,用包装试剂盒辅助转化到E.coliHB101感受态细胞中,并在含75 μg·mL-1潮霉素B的LB平板上筛选阳性菌落。挑取阳性菌落,摇菌,收集菌体,提取phAE159-AES噬菌粒。
1.2.3 制备M.smegmatis感受态细胞
挑取新鲜的M.smegmatis单克隆菌落接种于5 mL 7H9液体培养基中,37 ℃振荡培养至对数生长期(OD600约0.6)。以1∶100的比例将该菌液接种于100 mL 7H9培养基中,37 ℃振荡培养至OD600约0.6。将菌液冰浴1 h,然后于4 ℃,5 000 r·min-1离心10 min。收集菌体,用预先冰浴的10%无菌甘油将菌体洗涤2次,最后用10 mL预冷的10%的甘油重悬,每管200 μL分装并冻存于-80 ℃备用。
1.2.4 制备高滴度重组噬菌体
将phAE159-AES噬菌粒与M.smegmatis感受态细胞混匀,在BIO-RAD电转仪上用2 mm电转杯电击转化(电击参数:电压2.5 kV,电阻1 000 Ω,电容25 μF)。加入7H9培养基,在37 ℃培养箱中培养过夜。将此液体与顶层琼脂混匀并在7H10平板上铺板,30 ℃培养3 d,筛选噬菌体空斑。挑取平板上含有噬菌体的空斑到MP buffer中,4 ℃孵育过夜。将此含有噬菌体的液体与适量新鲜培养的M.smegmatismc2155混合,然后与适量顶层琼脂混匀,铺板,30 ℃培养3 d。将MP buffer加到含有噬菌体空斑的平板上,置于4 ℃过夜。用注射器收集平板上的液体,并用0.22 μm无菌滤菌器过滤除菌,获取高滴度的噬菌体,放4 ℃冰箱保存备用。
1.2.5MSMEG_0372基因敲除菌株的构建及鉴定
将高滴度的噬菌体与预先用MP buffer洗涤的对数期野生型M.smegmatis混匀,37 ℃孵育过夜。离心弃上清,补加适量7H9液体培养基,37 ℃孵育过夜。离心后收集细菌,并在含75 μg·mL-1潮霉素B的7H10平板上37 ℃培养3 d,筛选阳性菌落。挑取单克隆菌落,摇菌,收集菌体,提取细菌基因组DNA并用PCR鉴定。MSMEG_0372基因敲除菌株的验证引物见图1,左臂验证引物对的正向引物(LYZFP)位于在LFP引物匹配序列的上游,反向引物(LYZRP)位于sacB基因上;右臂验证引物对的正向引物(RYZFP)位于hyg基因上,反向引物(RYZRP)位于RRP引物匹配序列的下游。
1.2.6 构建pMV361-MSMEG_0372质粒及鉴定
以M.smegmatis的基因组DNA为模板(2 μL),以MSMEG_0372FP/MSMEG_0372RP为引物(F和R各0.5 μL),加入2×Taq Master mix(10 μL),补充无核酸酶水制备20 μL反应体系。PCR条件:95 ℃ 2 min;95 ℃ 20 s,55 ℃ 20 s,72 ℃ 105 s,共35个循环;72 ℃ 10 min。通过琼脂糖凝胶电泳对PCR扩增产物进行鉴定,回收MSMEG_0372基因片段。用限制性内切酶EcoRI和Hind III酶切MSMEG_0372基因片段和pMV361质粒,再用T4 DNA 连接酶连接起来,转化到E.coliDH5α感受态细胞中,并在含25 μg·mL-1卡那霉素的LB平板上筛选阳性菌落。挑取单克隆菌落,摇菌,收集菌体,提取质粒并通过DNA测序鉴定。MSMEG_0372基因片段被插入到pMV361 载体的多克隆位点上,pMV361-MSMEG_0372质粒的验证引物(JDFP/JDRP)位于该位点的上下游 100~200 bp处。
1.2.7MSMEG_0372基因回补菌株的构建与鉴定
制备MSMEG_0372基因敲除菌株感受态细胞(同野生型M.smegmatis感受态细胞的制备)。取适量pMV361-MSMEG_0372质粒电转化到MSMEG_0372基因敲除菌株感受态细胞中,并在含25 μg·mL-1卡那霉素和75 μg·mL-1潮霉素B的7H10平板筛选阳性菌落。挑取单克隆菌落,摇菌,收集菌体并煮沸裂解。以裂解液为模板,以JDFP/JDRP为引物进行PCR鉴定。
1.2.8MSMEG_0372基因过表达菌株的构建与鉴定
取适量pMV361-MSMEG_0372质粒电转化到野生型M.smegmatis感受态细胞中,并在含25 μg·mL-1卡那霉素的 7H10平板上筛选阳性菌落。挑取单克隆菌落,摇菌,收集菌体并煮沸裂解。以裂解液为模板,以JDFP/JDRP为引物进行PCR鉴定。
2 结果
2.1 成功构建p0004s-AES质粒
MSMEG_0372基因左、右臂扩增产物的琼脂糖凝胶电泳显示:左臂约为764 bp和右臂约为709 bp,与预期DNA片段大小一致(图2)。因为p0004s质粒含有4个限制性内切酶 Van91I酶切位点,酶切后产生了4个片段,回收其中的3.6 kbp和1.6 kbp片段。连接左、右臂(AES)以及p0004s质粒的两个大片段,构建的p0004s-AES质粒经测序显示:结果与预期一致,无突变。
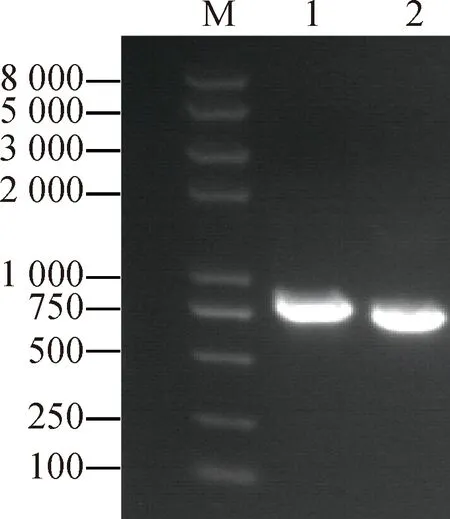
Lane 1:左臂; Lane 2:右臂;Lane M:DNA marker。图2 MSMEG_0372基因的左臂和右臂(AES)
2.2 M. smegmatis MSMEG_0372基因敲除菌株构建与鉴定
连接p0004s-AES和phAE159质粒酶切产物构建了phAE159-AES噬菌粒。将此噬菌粒转化到M.smegmatis感受态细胞中,筛选得到噬菌体空斑(含重组噬菌体)。用重组噬菌体侵染野生型M.smegmatis,得到高滴度的重组噬菌体。将高滴度的噬菌体与野生型M.smegmatis混合孵育,在37 ℃培养时,其自身DNA(phAE159-AES)与野生型M.smegmatis基因组DNA在AES处发生等位基因同源重组,完成MSMEG_0372基因的敲除。
2.2.1 “两片段法”PCR鉴定MSMEG_0372基因敲除菌株
“两片段法”PCR:分别使用左臂验证引物对(LYZFP/LYZRP)和右臂验证引物对(RYZFP/RYZRP)进行PCR。当以MSMEG_0372基因敲除菌株基因组DNA为模板时,能够扩增出约1 187 bp和1 128 bp大小的DNA片段,而当以野生型M.smegmatis基因组DNA为模板时则没有扩增产物。结果符合预期(图3)。
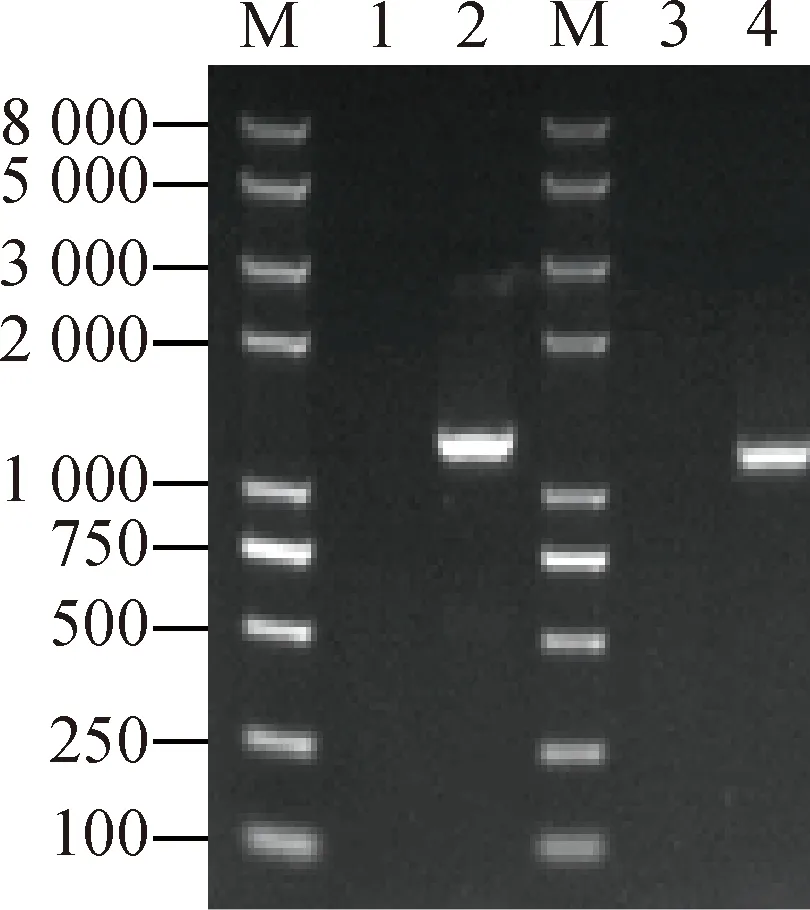
Lane 1:引物为LYZFP/LYZRP,模板为野生型M. smegmatis基因组DNA;Lane 2:引物为LYZFP/LYZRP,模板为MSMEG_0372 基因敲除菌株基因组DNA;Lane3: 引物为RYZFP/RYZRP,模板为野生型M. smegmatis基因组DNA;Lane 4:引物为RYZFP/RYZRP,模板为MSMEG_0372 基因敲除菌株基因组DNA;Lane M:DNA Marker。图3 “两片段法”PCR 鉴定 MSMEG_0372 基因敲除菌株
2.2.2 “全长法”PCR鉴定MSMEG_0372基因敲除菌株
“全长法”PCR:组合使用左臂验证正向引物和右臂验证反向引物(LYZFP/RYZRP)进行PCR。当以MSMEG_0372基因敲除菌株基因组DNA为模板时,能够扩增出约5 616 bp大小的DNA片段,而当以野生型M.smegmatis基因组DNA为模板时,能够扩增得到约2 772 bp大小的DNA片段。结果符合预期(图4)。两种PCR的鉴定结果表明:MSMEG_0372基因敲除菌株 (mc2155 ΔMSMEG_0372) 构建成功。
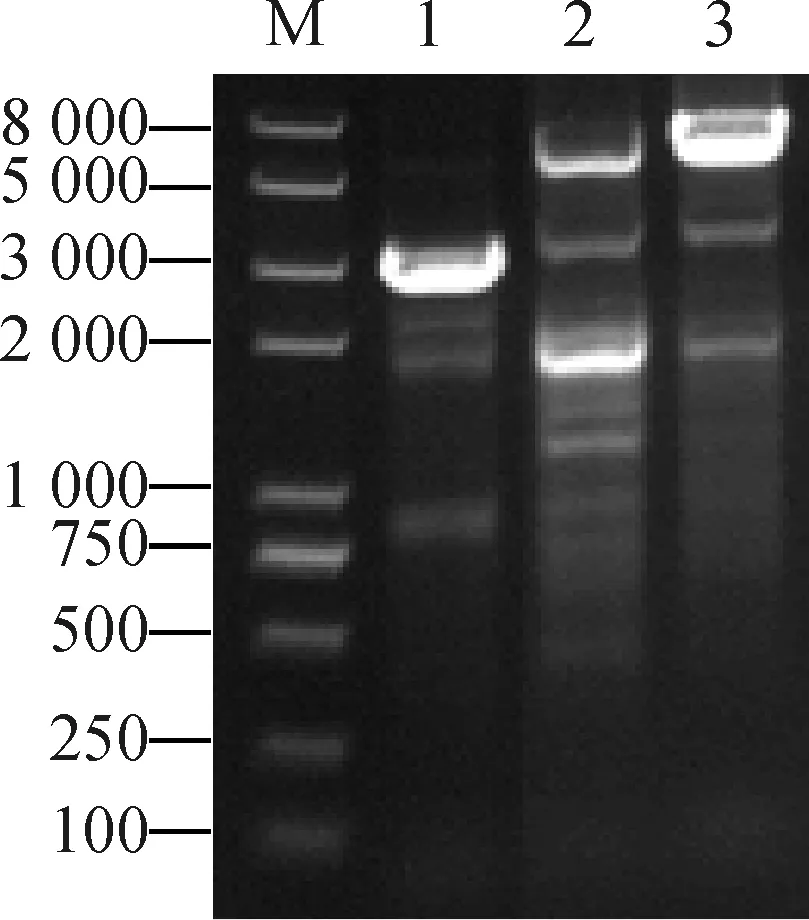
Lane 1:引物为LYZFP/RYZRP,模板为野生型M. smegmatis基因组DNA;Lane2-3:引物为LYZFP/RYZRP,模板为MSMEG_0372基因敲除菌株基因组DNA;Lane M:DNA Marker。图4 “全长法”PCR 鉴定 MSMEG_0372 基因敲除菌株
2.3 MSMEG_0372基因回补菌株的构建与鉴定结果
将pMV361-MSMEG_0372质粒导入MSMEG_0372基因敲除菌株,得到MSMEG_0372基因回补菌株的阳性菌落。以细菌裂解液为模板,以JDFP/JDRP为引物进行PCR,电泳结果显示:片段大小约1 651 bp,符合预期,MSMEG_0372基因回补菌株(mc2155 ΔMSMEG_0372:pMV361-MSMEG_0372)构建成功(图5)。
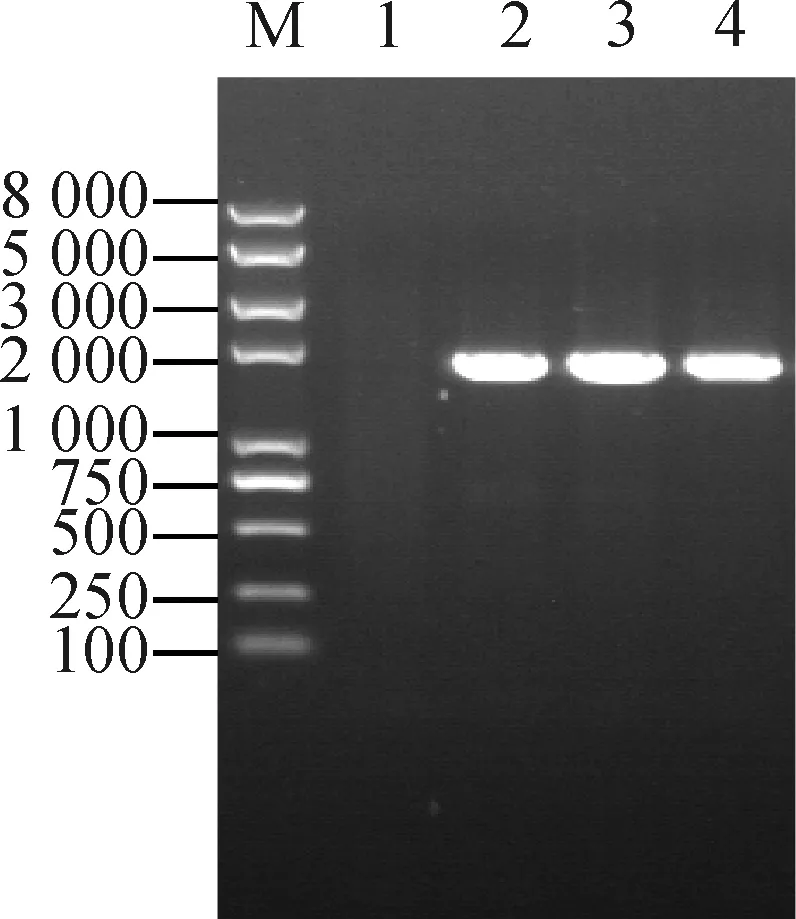
Lane1:引物为JDFP/JDRP,模板为野生型M. smegmatis裂解液;Lane 2-4:引物为JDFP/JDRP,模板为MSMEG_0372 基因回补菌株裂解液;Lane M:DNA Marker。图5 PCR 鉴定 MSMEG_0372 基因回补表达菌株
2.4 M. smegmatis MSMEG_0372基因过表达菌株的鉴定结果
M.smegmatisMSMEG_0372基因过表达菌株的鉴定结果见图6。
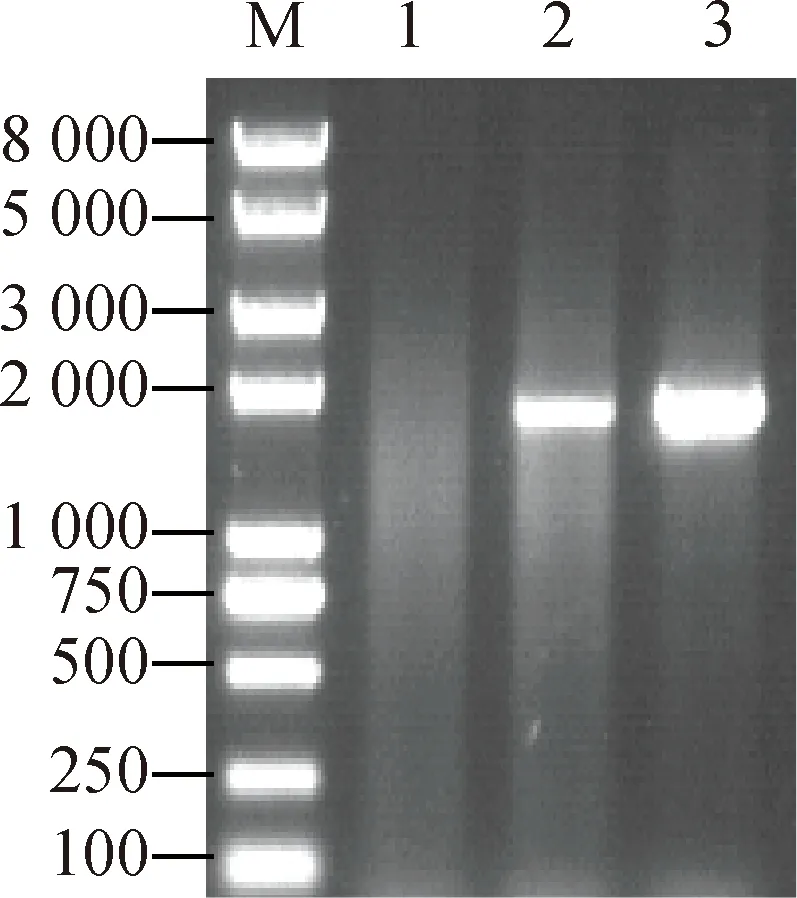
Lane 1:引物为JDFP/JDRP,模板为野生型M. smegmatis裂解液;Lane 2-3:引物为JDFP/JDRP,模板为MSMEG_0372 基因过表达菌株裂解液;Lane M:DNA Marker图6 PCR 鉴定 MSMEG_0372 基因过表达菌株
将pMV361-MSMEG_0372质粒导入野生型M.smegmatis,得到MSMEG_0372基因过表达菌株的阳性菌落。以细菌裂解液为模板,以JDFP/JDRP为引物进行PCR,电泳结果显示:片段大小约1 651 bp,符合预期,MSMEG_0372基因过表达菌株(mc2155:pMV361-MSMEG_0372)构建成功(图6)。
3 讨论
近年来,耐药TB已经成为TB防控领域的一大挑战[8]。随着M.tuberculosis基因组测序的完成,人们开始更深入地研究基因的功能[9],从而筛选靶点蛋白并研发抗结核新药[10]。通过构建基因敲除、回补与过表达菌株,比较分析菌株间的差异,我们可以精确地研究目的基因的功能。M.smegmatis是一种从梅毒患者的硬下疳中分离得到的腐生菌,生长迅速且为非致病菌,在生物安全一级标准(Biosafety Level 1,BSL-1)的实验室即可操作。同时,M.smegmatis与M.tuberculosis细胞结构相似,同源性高,是一种理想的M.tuberculosis模式菌[11]。
与其他细菌相比,分枝杆菌细胞壁厚,脂质含量高,生长缓慢,且基因组同源重组率低,这使得分枝杆菌基因敲除菌株的构建仍然是细菌遗传操作中的一大难点[12]。目前,M.tuberculosis基因敲除的方法主要有:基于噬菌体的特异性转导法、线性DNA片段法、反选择标记法、成簇规律性间隔短回文重复(Clustered regularly interspaced short palindromic repeats, CRISPR)法以及 Bxb1整合酶靶向寡核苷酸介导的基因重组(oligonucleotide-mediated recombineering followed by Bxb1 integrase targeting, ORBIT)[13]。特异性转导法转化率和敲除效率都很高。线性DNA片段基因敲除法提高了重组率,但错配率升高[13]。反选择标记基因敲除法依赖于特定基因,失活率高[13]。CRISPR/Cas是细菌免疫系统的重要成分[14],能够精确识别及剪切特异性DNA序列[15]。CRISPR基因敲除法操作简单,突变率高,特别适于探索功能未知的基因[13]。ORBIT法则结合了Che9-RecT和 Bxb1噬菌体整合酶系统,可以同时创建缺失、插入或融合的文库,应用前景广阔[13]。
在本实验中,我们采用特异性噬菌体转导法构建了MSMEG_0372基因敲除菌株。我们构建了由分枝杆菌噬菌体DNA(phAE159)和MSMEG_0372基因的左右臂(含hyg基因盒)组成的噬菌粒—phAE159-AES。这种噬菌粒在分枝杆菌中以噬菌体的方式进行复制,而在E.coli中则以质粒的方式进行复制,当其在分枝杆菌和E.coli进行穿梭时可以促进其携带的DNA发生转移[13]。phAE159是一种温敏型噬菌体,在30 ℃ 裂解生长,在37 ℃ 溶原生长[16]。所以,在30 ℃ 培养phAE159-AES重组噬菌体时,其可以在M.smegmatis中扩增释放,而在37 ℃培养时,其可以将重组噬菌体的DNA(噬菌粒phAE159-AES)整合到M.smegmatis基因组DNA中,噬菌粒的AES (MSMEG_0372基因左、右 臂)与野生型M.smegmatis基因组DNA中的等位基因发生同源重组,位于AES中间的MSMEG_0372基因也被重组噬菌粒上含hyg基因盒的DNA片段所替换,从而完成基因敲除。经过潮霉素B抗性平板的筛选,得到MSMEG_0372基因敲除菌株的阳性菌落。通过上、下游验证引物对的不同组合,我们用“两片段法”和“全长法”PCR,更加全面、准确地鉴定了该菌株。与另一常用噬菌体phAE87相比,phAE159容量更大,克隆能力更强,DNA的传递效率也更高[17]。同时,由于本实验主要在E.coli和快速生长分枝杆菌M.smegmatis中进行,所以本实验还具有周期短的特点。特异性转导法广泛适用于各种分枝杆菌[13],其不足之处在于噬菌粒的构建相对复杂[17]。
在本实验中,我们将pMV361-MSMEG_0372质粒分别导入MSMEG_0372基因敲除菌株和野生型M.smegmatis,构建了MSMEG_0372基因回补和过表达菌株。pMV361是一种含有M.tuberculosis热休克蛋白60(hsp60)基因启动子的大肠杆菌-分枝杆菌穿梭整合质粒,可以促进插入基因的表达。基因敲除可以反向验证MSMEG_0372基因的功能,基因过表达可以正向验证该基因功能,而基因回补在理论上可以回复基因敲除的改变。总之,MSMEG_0372基因敲除、回补与过表达菌株的成功构建,为研究该基因在M.smegmatis中的功能奠定了良好的基础。未来可以进一步研究FabG4在分枝杆菌 Ⅱ 型脂肪酸合成(FAS-Ⅱ)途径中的作用、参与生物膜形成的机制,评价其作为分枝杆菌生物膜标记物的可行性和抗结核药物作用靶点的价值,这对 TB 的诊断、治疗和预防具有重要意义。
