Hepatic transcriptome profiling reveals early signatures associated with disease transition from non-alcoholic steatosis to steatohepatitis☆
2023-01-09NncyMgeeForknAhmedNtlieEpplerElizbethJonesPriynkGhoshLilyHeYuxiZhng
Nncy Mgee ,Forkn Ahmed ,Ntlie Eppler ,Elizbeth Jones ,Priynk Ghosh ,Lily He ,Yuxi Zhng ,b,*
a Department of Pharmacology,Toxicology and Therapeutics,University of Kansas Medical Center,Kansas City,KS,USA
b Liver Center,University of Kansas,Kansas City,KS,USA
Keywords:Liver Transcriptome Steatosis Inflammation Fibrosis Non-alcoholic steatohepatitis (NASH)
ABSTRACT Background and aim: Non-alcoholic fatty liver disease (NAFLD) is becoming a leading cause of chronic liver disease worldwide.The molecular events that influence disease progression from non-alcoholic fatty liver (NAFL) to aggressive non-alcoholic steatohepatitis (NASH) remain incompletely understood,leading to lack of mechanism-based targeted treatment options for NASH.This study aims to identify early signatures associated with disease progression from NAFL to NASH in mice and humans.Materials and methods: Male C57BL/6J mice were fed a high-fat,-cholesterol,and -fructose (HFCF) diet for up to 9 months.The extent of steatosis,inflammation,and fibrosis was evaluated in liver tissues.Total RNA sequencing (RNA-seq) was conducted to determine liver transcriptomic changes.Results: After being fed the HFCF diet,mice sequentially developed steatosis,early steatohepatitis,steatohepatitis with fibrosis,and eventually spontaneous liver tumor.Hepatic RNA-seq revealed that the key signatures during steatosis progression to early steatohepatitis were pathways related to extracellular matrix organization and immune responses such as T cell migration,arginine biosynthesis,C-type lectin receptor signaling,and cytokine-cytokine receptor interaction.Genes regulated by transcription factors forkhead box M1 (FOXM1) and negative elongation factor complex member E (NELFE) were significantly altered during disease progression.This phenomenon was also observed in patients with NASH.Conclusions: In summary,we identified early signatures associated with disease progression from NAFL to early NASH in a mouse model that recapitulated key metabolic,histologic,and transcriptomic changes seen in humans.The findings from our study may shed light on the development of novel preventative,diagnostic,and therapeutic strategies for NASH.
1.Introduction
Non-alcoholic fatty liver disease(NAFLD)is becoming a leading cause of chronic liver disease worldwide due to the increasing prevalence of obesity,hyperlipidemia,and diabetes.1NAFLD is a disease spectrum,ranging from simple steatosis or non-alcoholic fatty liver (NAFL) to the more aggressive non-alcoholic steatohepatitis (NASH).NAFL is generally considered a relatively benign condition;however,approximately 20-30% of these patients develop NASH that is characterized by steatosis with hepatocyte ballooning (cell death),liver inflammation with or without fibrosis,which can further progress to irreversible cirrhosis and hepatocellular carcinoma(HCC)without effective treatments.2-4To date,much progress has been made to understand the pathogenesis of NAFLD;however,the molecular events that influence the transition of NAFL to NASH remain poorly understood,leading to a lack of mechanism-based targeted treatment options for NASH.Thus,understanding the molecular machinery that causes the transition of simple steatosis to NASH is essential to develop effective prevention and therapeutic strategies for this unresolved disease.
Several dietary animal models have been used to characterize the pathogenesis of NAFLD,including animals fed methionine choline-deficient(MCD)diet or Western-type diets with an excess of saturated fats,trans fats,and cholesterol;either individual use or in combination with sugars such as fructose and sucrose.5-7Ideally,a preclinical animal model for NASH should be associated with the same risk factors seen in humans such as obesity,insulin resistance,dyslipidemia,and endocrine dysfunction.The animal model should also match NASH in humans with respect to histological characteristics(steatosis,hepatocyte damage,inflammation,and fibrosis)as well as alterations in gene expression signatures and signaling pathways relevant to humans.Mice fed an MCD diet develop steatosis,inflammation and fibrosis;however,this model lacks features of metabolic syndrome such as obesity,dyslipidemia,and insulin resistance.6Mice fed high-fat and high-cholesterol diet feature obesity,insulin resistance,dyslipidemia,and steatosis with liver inflammation,but do not develop liver fibrosis.6Recently,a diet enriched in fat,cholesterol,and fructose has been implicated in the development of obesity and NASH in humans.8,9Similarly,highfat diets enriched in fructose and various amounts of cholesterol(0.2%or 2.0%)effectively induce NASH in mice.5,10-14In such a diet,excess fat alone contributes to the development of hepatic steatosis while the addition of fructose and cholesterol predispose animals to hepatocyte damage,inflammation and fibrosis.
Over the past decades,bioinformatics tools and techniques such as genome-wide associated studies (GWAS) and transcriptomewide analysis of gene expression by RNA sequencing (RNA-seq)have been widely employed as powerful tools to investigate molecular mechanisms implicated in NASH development in humans and mouse models.15-18An earlier study compared hepatic messenger RNA (mRNA) expression profiles between NAFLD patients and various mouse models of NAFLD,including mice on a high-fat diet with or without fructose,mice on an MCD diet,mice on a high-fat diet given streptozotocin,and mice with disruption of hepatic phosphatase and tensin homolog (Pten) gene.16Surprisingly,the comparisons yielded very few overlaps between human NASH and mouse models of NASH,indicating that NASH development in mouse models does not significantly correlate with NASH developed in humans at the transcriptome level.However,a recent study that employed RNA-seq exploring hepatic mRNA expression profile in a mouse NAFLD model developed in inbred isogenic C57BL/6J×129S1/SvlmJ mice fed a cholesterol-enriched HFD with consumption of a high fructose and glucose water (termed DIAMOND mice) has demonstrated that the DIAMOND model recapitulated transcriptomic and cell-signaling changes seen in humans with progressive NASH,suggesting some similarities between human NASH and mouse model of NASH at both histological and transcriptomic levels.19
In the current study,we fed mice a high-fat diet enriched in cholesterol and fructose(HFCF,40 kcal%fat,2%cholesterol,20 kcal%fructose) for up to 9 months and monitored NAFLD development.We found that the HFCF-fed mice sequentially developed steatosis(1 month),borderline steatohepatitis (3 months),definite steatohepatitis (5 months),steatohepatitis with progressive fibrosis (7 months onwards),and spontaneous liver tumor (9 months).To pursue an unbiased investigation of early events during the transition of steatosis to borderline steatohepatitis,we conducted RNAseq analysis of livers from 1 month to 3 months HFCF-fed mice.The hepatic transcriptome profiling comparison revealed that the early signatures associated with disease progression from steatosis to borderline steatohepatitis were extracellular matrix (ECM) organization and immune responses such as T cell migration,arginine biosynthesis,C-type lectin receptor signaling,and cytokinecytokine receptor interaction.Genes regulated by transcription factors forkhead box M1 (Foxm1) and negative elongation factor complex member E (Nelfe) were significantly altered during the disease transition.This phenomenon was not only observed in NAFL transition to early NASH in mouse model but also observed in disease progression of NAFLD patients.In summary,we characterized NAFLD development in a mouse model that recapitulated key metabolic,histologic,and transcriptomic changes seen in humans.In addition,transcriptome-wide analysis of gene expression by RNA-seq identified the early signatures associated with disease transition from steatosis to borderline steatohepatitis.The findings from our study may pave the way to identify useful diagnostic biomarkers of NASH and aid in developing novel preventative and therapeutic strategies for NASH.
2.Materials and methods
2.1.Animal studies
C57BL/6J (#000664) mice were purchased from Jackson Laboratory(Bar Harbor,ME,USA).Mice were maintained in a 12 h light/dark cycle (light on 6 a.m.to 6 p.m.),temperature-controlled(22-23°C),and virus-free facility with free access to food and water.Experiments on mice were performed on males at the age of 8-10 weeks (n=5/group).For dietary NAFLD models,C57BL/6J mice were either placed on a normal chow diet(D12450J,10 kcal%fat,Research Diet,New Brunswick,NJ,USA)or a diet enriched in fat,cholesterol,and fructose (D09100301,40 kcal% fat mostly from trans fat Primex,2% cholesterol,20 kcal% fructose,Research Diet,New Brunswick,NJ,USA) for up to 9 months.Serum and liver samples were collected after mice were fasted for 16 h.All experiments were performed in accordance with relevant guidelines and regulations approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Kansas Medical Center.
2.2.Glucose tolerance test (GTT) and insulin tolerance test (ITT)
For GTT assay,mice were fasted for 16 h,followed by intraperitoneal injection of glucose (Sigma,St.Louis,MO,USA) at 2 g/kg body weight.Blood was collected by minor tail clipping and glucose levels were determined before and at 30,90,120,and 180 min after glucose administration.In ITT assay,mice were fasted for 4 h from 8 a.m.to 12 p.m.,followed by intraperitoneal injection of insulin(Sigma,St.Louis,MO,USA)at 0.75 U/kg body weight.Blood glucose was measured before and at 30,60,90,and 120 min post insulin injection.
2.3.Serum measurements
Blood samples were collected from anesthetized animals via cardiac puncture.Various kits from Pointe Scientific (Canton,MI,USA)were used to determine serum levels of triglycerides(T7532),total cholesterol (C7510),glucose (G7521),aspartate aminotransferase(AST) (A7561),and alanine aminotransferase (ALT) (A7526).
2.4.Liver histology
Fresh liver tissues were fixed with 10% formalin (Fisher Scientific,Waltham,MA,USA).Paraffin sections at 4 μm were stained with hematoxylin and eosin (H&E) to assess liver histology.A pathologist conducted blinded histopathology evaluation and scored the severity of steatosis,inflammation,cell death,and fibrosis according to the NASH Clinical Research Network (CRN)criteria.19,20In brief,the amount of steatosis was scored as 0(<5%),1 (5-33%),2 (>33-66%) and 3 (>66%).Hepatic cell death was scored as 0 (none),1 (few) and 2 (many cells).Foci of lobular inflammation were scored as 0(no foci),1(<2 foci),2(2-4 foci)and 3(>4 foci).Fibrosis was scored as 0(no fibrosis),1(perisinusoidal or periportal fibrosis),2(perisinusoidal and portal/periportal fibrosis),3 (bridging fibrosis) and 4 (cirrhosis).
2.5.Oil red O staining of lipids
Fresh liver tissues were immediately embedded in Tissue-Tek O.C.T.compound (Sakura Finetek,Torrance,CA,USA).Frozen sections were cut at 8 μm and fixed with 10% formalin,followed by stained with Oil Red O(Sigma,St.Louis,MO,USA)and hematoxylin.Images were acquired with a BX60 microscope (Olympus,Lake Success,NY,USA).
2.6.Immunohistochemistry
Antibodies used for immunohistochemistry staining,including F4/80 (70076),CD4 (25229),CD8 (98941),CD19 (90176),CD11C(97585),Ly-6G (87048),and proliferating cell nuclear antigen(PCNA) (13110) were purchased from Cell Signaling (Danvers,MA,USA).In brief,paraffin sections at 4 μm were rehydrated and incubated with 0.3% H2O2for 15 min to block endogenous peroxidase activity.Antigen retrieval was acquired by heat-induced antigen retrieval using citrate buffer (pH 6.0) for 10 min.Slides were then treated with 10% normal serum for 30 min,followed by incubation with primary antibody overnight at 4°C.ImmPRESS peroxidase polymer detection kits (Vector Laboratories,Newark,CA,USA) and ImmPACT 3,3′-diaminobenzidine (DAB) peroxidase substrate(Vector Laboratories,Newark,CA,USA)were used for the final detection.Sections were then counterstained with hematoxylin,dehydrated,cleared,and mounted.Images were acquired with a BX60 microscope.TUNEL staining for detection of cell death in the liver was performed using an in-situ cell death detection kitalkaline phosphatase (Sigma,St.Louis,MO,USA) according to the manufacturer's suggestions.
2.7.Picrosirius red staining of liver fibrosis
Paraffin sections at 4 μm were rehydrated and incubated in 0.1%Sirius red F3B (Sigma,St.Louis,MO,USA) containing saturated picric acid for 1 h.After washing three times in 0.5% glacial acetic acid,sections were briefly dehydrated,cleared,and mounted.Images were acquired with a BX60 microscope.
2.8.Hepatic triglycerides and cholesterol content
The method has been described previously.14In brief,100 mg of liver tissues were homogenized in 300 μL of chloroform:methanol(1:2 in volume)for 2 min,followed by a second homogenization for 30 s with an addition of 300 μL chloroform.The homogenates were then mixed with 100 μL of H2O and homogenized again for 30 s.Lipid layer(~600 μL)was separated via centrifugation at 800×gfor 10 min at room temperature.The lipid extract was then dried using nitrogen gas and suspended in 300 μL of 5% Triton X-100 in phosphate buffered saline (PBS) (pH 7.4).Lipid contents were determined using respective kit for triglycerides and cholesterol(Pointe Scientific,Canton,MI,USA).The hepatic triglycerides or cholesterol contents were defined as μg of triglycerides or cholesterol per mg of liver tissue.
2.9.Liver hydroxyproline assay
Liver tissues (10 mg) were homogenized in 100 μL of H2O and mixed with 100 μL of 12 M HCl,followed by acid hydrolysis at 120°C for 3 h.The homogenates were then centrifuged at 10,000×gfor 10 min.Aliquots of the hydrolyzed samples (10 μL) were incubated with 100 μL of chloramine T solution(1.27%chloramine T and 10% isopropanol in acetate-citrate buffer,pH 6.0) at room temperature for 25 min,followed by a second incubation with 100 μL of Ehrlich's solution(Sigma,St.Louis,MO,USA)at 60°C for 35 min.A plate reader measured absorbance at 550 nm.The hepatic collagen content was defined as μg of collagen per mg of liver tissue.
2.10.RNA sequencing and data analysis
Total RNA for Illumina sequencing was extracted from mouse liver tissues using a Direct-zol RNA kit (Zymo Research,Irvine,CA,USA).DNA was removed by treating samples with RNase-free DNase.The purity,concentration and integrity of RNA were examined by a NanoDrop 1000 spectrophotometer(Thermo Fisher,Waltham,MA,USA) and an Agilent Bioanalyzer 2100 system(Agilent Technologies,Santa Clara,CA,USA).Three biological replicates from mice fed either a chow or HFCF diet for 1 month or 3 months were submitted for RNA sequencing.The RNA integrity number(RIN) values of all samples used for RNA-seq were >6.0.The high throughput Genomics Core at the Huntsman Cancer Institute at the University of Utah prepared cDNA library using the Illumina TruSeq stranded RNA kit with Ribo-Zero Gold.Fifty-cycle single-read sequencing was performed with an Illumina HiSeq 2500(Illumina,San Diego,CA,USA) and reads were aligned to a mouse reference sequence genome mm10 using the Novalign short-read alignment software.Sample reads were visualized,and the differentially expressed genes (DEGs) were identified based on the logtransformed false discovery rate (FDR) of >1.3 and ≥± 1.5-fold change in expression relative to controls using the USeq application as previously described.21All RNA-seq data generated in this work were deposited into the Gene Expression Omnibus (GEO)database (GSE135050).Gene Ontology (GO) term enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses using DEGs were performed in Enrichr web server to identify enriched biological functions and pathways.22,23Enrichr is freely available at: http://amp.pharm.mssm.edu/Enrichr.GO terms and KEGG pathways withP-value <0.05 were considered significantly different.An Enrichr combined score calculated based on adjustedP-value and Z score was used to select top enriched GO terms and KEGG pathways.
2.11.Real-time quantitative polymerase chain reaction (qPCR)
qPCR was carried out using the SYBR Green PCR master mix(Applied Biosystems,Waltham,MA,USA).qPCR primer sequences for peroxisome proliferator activated receptor gamma (Pparg),CD antigen 36 (Cd36),cell death-inducing DFFA-like effector c (Cidec,also calledFsp27),acetyl-Coenzyme A carboxylase alpha (Acaca,also calledAcc1),fatty acid synthase (Fasn),stearoyl-Coenzyme A desaturase 1 (Scd1),peroxisome proliferator activated receptor alpha (Ppara),carnitine palmitoyltransferase 1 (Cpt1),apolipoprotein B (ApoB),microsomal triglyceride transfer protein (Mttp),tumor necrosis factor alpha (Tnfα),chemokine (C-C motif) ligand 2(Ccl2),nitric oxide synthase 2(Nos2),CD163,arginase-1(Agr1),and collagen 1 alpha 1 (Col1a1) were previously published.14The amount of PCR products was measured by cycle threshold (Ct)values,and the relative ratio of specific genes to housekeeping gene hypoxanthine guanine phosphoribosyl transferase 1 (Hprt1) was calculated and presented as a fold change in the tested group relative to the control group.
2.12.Human NAFLD microarray dataset
The microarray dataset of human livers including 14 normal,14 NAFL,and 18 NASH is accessible in NCBI GEO database GSE48452.
2.13.Statistical analysis
Quantitative data were presented as the mean±standard error of the mean (SEM).The statistical difference in data obtained between the two groups was determined by unpaired two-tailed Student'st-test.Multiple groups were compared by one-way analysis of variance (ANOVA) and followed by Duncan's test.Statistical significance was accepted within 95% confidence limits.
3.Results
3.1.HFCF-fed mice develop obesity,dyslipidemia,and insulin resistance
To develop a mouse model with disease progression from NAFL to NASH,C57BL/6J mice were fed a high-fat diet enriched in cholesterol and fructose for up to 9 months and samples were collected at 1,3,5,7,and 9 months post diet administration(Fig.1A).Mice gained weight rapidly on the HFCF diet with the maximum weight gain at the 7 months,after which the growth remained stable (Fig.1B).Paralleling body weight gains,both liver weight and liver-to-body weight ratio increased rapidly in the HFCF-fed mice,becoming significantly different from the chow-fed mice after 3 months of feeding (Fig.1C).Meanwhile,HFCF diet feeding led to a persistent increase in liver injury markers ALT and AST (Fig.1D).Parallelly,mice fed HFCF diet increased fasting cholesterol from the 3-month throughout the experimental period(Fig.1E).In contrast,mice fed 1 month of HFCF diet developed hypertriglyceridemia,after which serum triglycerides levels declined to values like chow-fed mice(Fig.1E).Moreover,HFCF-fed mice developed glucose intolerance and insulin resistance at the 3-month post diet feeding (Fig.1F and G).Collectively,these results indicate that HFCF-fed mice developed obesity,dyslipidemia,and insulin resistance.
3.2.HFCF feeding induces liver steatosis
Mice fed HFCF diet rapidly developed both microvesicular and macrovesicular steatosis after 1 month of diet administration(Fig.2A).The latter predominated from the 3-month onwards and were seen commonly in zone 1.Liver steatosis was also confirmed by the Oil red O staining(Fig.2A)and quantification of triglyceride and cholesterol from liver lipid extraction(Fig.2B).
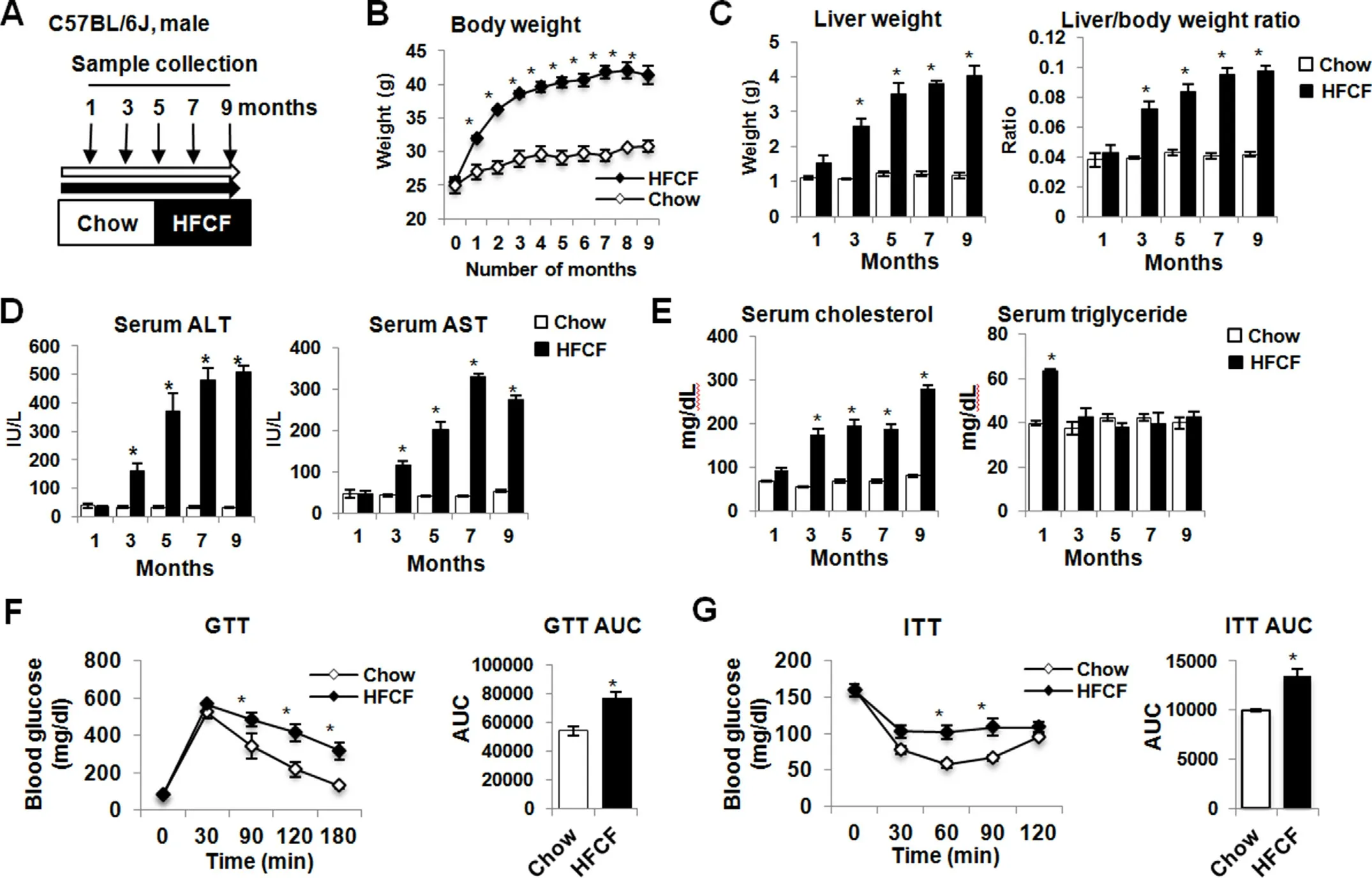
Fig.1.HFCF-fed mice develop obesity,dyslipidemia,insulin resistance,and liver injury.(A)Experimental design showing diet feeding and time schedule for sample collection.C57BL/6J mice were fed chow or an HFCF diet for up to 9 months.(B) Body weight change over time.(C) Liver weight and liver-to-body weight ratio.(D) Serum ALT and AST.(E)Fasting serum levels of cholesterol and triglycerides.(F)GTT in mice on diet for 3 months.(G)ITT in mice on diet for 3 months.Data are expressed as the mean±SEM for 5 mice per group.*P <0.05 versus chow controls.Abbreviations: ALT,alanine aminotransferase;AST,aspartate aminotransferase;AUC,area under curve;GTT,glucose tolerance test;HFCF,high-fat,cholesterol,and fructose;ITT,insulin tolerance test;SEM,standard error of the mean.
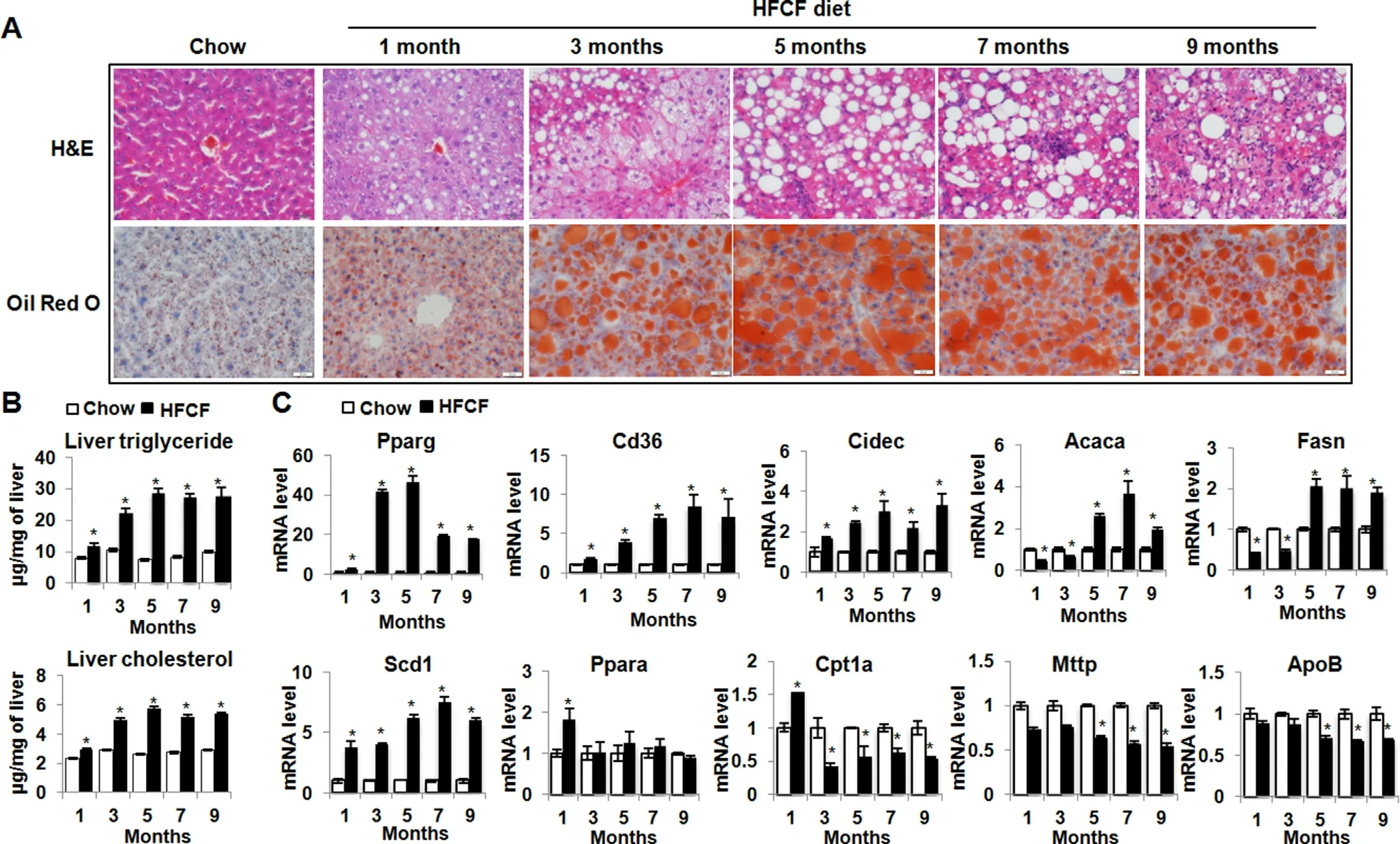
Fig.2.HFCF-fed mice develop liver steatosis.Two-month-old C57BL/6J male mice were fed chow or an HFCF diet for up to 9 months.(A) Representative images of liver sections stained with H&E or oil red O.Original magnification,× 40.(B) Liver triglycerides and cholesterol content.(C) The relative mRNA levels of genes involved in lipid metabolism were determined by qPCR.Data presented in Fig.B and C are expressed as the mean±SEM for 5 mice per group.*P<0.05 versus chow controls.Abbreviations:Acaca,acetyl-Coenzyme A carboxylase alpha;ApoB,apolipoprotein B;Cd36,CD antigen 36;Cidec,cell death-inducing DFFA-like effector c;Cpt1,carnitine palmitoyltransferase 1;Fasn,fatty acid synthase;H&E,hematoxylin and eosin;HFCF,high-fat,-cholesterol,and-fructose;Mttp,microsomal triglyceride transfer protein;Ppara,peroxisome proliferator activated receptor alpha;Pparg,peroxisome proliferator activated receptor gamma;Scd1,stearoyl-Coenzyme A desaturase 1;SEM,standard error of the mean.
Pparg,a master transcriptional regulator of lipid uptake and storage,sustained an increase in the livers of HFCF-fed mice(Fig.2C).In parallel,Pparg-regulated direct targets such as fatty acid transporterCd36and lipid droplet membrane protein geneCidec,also calledFsp27,were significantly increased at all time points throughout the 9-month feeding period(Fig.2C).In contrast,genes involved inde novolipogenesis (DNL),includingAcaca,also calledAcc1,andFasnwere decreased in HFCF-fed mice within 3 months of introducing diet,but increased precipitously at the 5-month and thereafter remained 2-to 4-fold elevation (Fig.2C).Scd1,a ratelimiting enzyme for converting saturated fatty acids (SFAs) to monounsaturated fatty acids (MUFAs),sustained increased expression levels throughout the 9-month feeding period(Fig.2C).Ppara,a key transcriptional regulator of genes involved in peroxisomal and mitochondrial beta-oxidation,was transiently induced in HFCF-fed mice within 1 month of diet administration,and thereafter decreased to the levels noted in the chow-fed groups(Fig.2C).Moreover,Cpt1,a direct target ofPparaand transports fatty acids (FAs) through the inner mitochondrial membrane for beta-oxidation,was significantly increased in the 1-month HFCFfed group but thereafter declined significantly throughout the 9 months feeding period (Fig.2C),suggesting that short-term HFCF feeding increases fatty acid oxidation,but long-term feeding represses it.ApoBandMttp,two critical genes involved in the secretion of very-low-density lipoprotein (VLDL),were unchanged during the first 3 months of HFCF diet administration,but thereafter decreased significantly (Fig.2C),consistent with changes of serum triglyceride levels observed in the HFCF-fed animals(Fig.1E).Taken together,the above data suggest that HFCF feeding induces liver steatosis and coordinately alters expression of genes involved in fatty acids uptake,biosynthesis,storage,beta-oxidation,and lipid secretion.
3.3.HFCF diet feeding triggers hepatic innate and adaptive immune responses
NASH pathogenesis involves not only the innate immunity mediated by macrophages,dendritic cells,and neutrophils,but also the adaptive immunity mediated by T and B cells.24To characterize immune cell types during NAFLD development,liver sections of HFCF-fed groups were stained with macrophage marker F4/80,dendritic cell marker CD11C,neutrophil marker Ly6G,T cell markers CD4 and CD8,and B cell marker CD19.Macrophage recruitment and activation are key elements in NASH.25Consistently,we found a progressive increase in hepatic macrophages in HFCF-fed groups,starting at the 3-month following the diet(Fig.3A).Dendritic cells are antigen-presenting cells that initiate potent adaptive immune responses.26Earlier studies revealed intrahepatic expansion and maturing of dendritic cells during NASH development.27,28In line with these observations,an expansion of CD11C+dendritic cells was observed in the 3-month HFCF-fed group,which remained at an elevated level for the duration of disease progression (Fig.3A).Neutrophil infiltration was observed in HFCF-fed groups after the 5-month diet feeding(Fig.3A).We next examined immune cells in adaptive immunity.Fig.3A showed a progressive increase in CD4+T cells and CD8+T cells in HFCF-fed groups after the 5-month diet administration.In contrast,the hepatic CD19+B cell infiltration was obvious in the 7-month HFCF-fed group (Fig.3A).Taken together,the above data confirmed the involvement of both innate and adaptive immunity in NAFLD development.
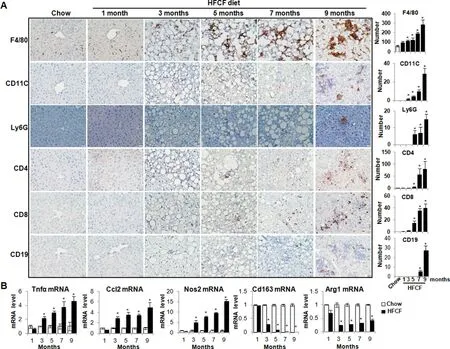
Fig.3.HFCF diet feeding triggers hepatic innate and adaptive immune responses.Two-month-old C57BL/6J male mice were fed chow or an HFCF diet for up to 9 months.(A)Left panel,representative images of liver sections stained with various immune cell markers.Right panel,quantifications of positive stained cells per view.Original magnification,×40.(B)The relative mRNA levels of genes involved in inflammation were determined by qPCR.Data are expressed as mean±SEM for 5 mice per group.*P<0.05 versus chow controls.Abbreviations:Agr1,arginase-1;Ccl2,chemokine(C-C motif)ligand 2;HFCF,high-fat,cholesterol,and fructose;Nos2,nitric oxide synthase 2;qPCR,quantitative polymerase chain reaction;SEM,standard error of the mean;Tnfα,tumor necrosis factor alpha.
At the molecular levels,the expression ofTnfα andCcl2,the two most well-recognized pro-inflammatory chemoattractants to monocytes and macrophages,were both significantly increased in the 3-month HFCF-fed group and sustained increased levels over the time of HFCF diet administration (Fig.3B).In contrast to the progressive increase in M1 macrophage markerNos2,M2 macrophage markers CD163 andAgr1were both significantly decreased in HFCF-fed groups after the 3-month diet feeding,suggesting an overall predominance of pro-inflammatory M1 like macrophages in the livers of HFCF-fed groups (Fig.3B).
3.4.HFCF diet feeding results in hepatic cell death, fibrosis,and tumor development
We next assessed the extent of cell death and fibrosis in HFCFfed groups.In consistent with elevated serum AST and ALT levels(Fig.1D),TUNEL staining detected an increase in hepatic cell death in HFCF-fed groups,starting at the 3-month after diet administration,and remained at an elevated level throughout the 9 months feeding period(Fig.4A).In parallel,liver fibrosis started to increase in HFCF-fed groups after the 3-month feeding,evidenced by an increase in collagen deposition detected by both Picrosirius red staining (Fig.4A) and quantification of hepatic hydroxyproline extraction (Fig.4B).Similarly,fibrosis markerCol1a1increased in the 3-month HFCF-fed group and remained an increased expression throughout the disease progression (Fig.4C).Finally,spontaneous hepatic tumors developed in 20% (2/10) of the mice maintained on the HFCF diet for 9 months (Fig.4D).
We then calculated the NAFLD activity score (NAS) in HFCF-fed mice over the course of diet administration.NAS increased at the 1-month HFCF-fed group and remained significantly higher than the chow-fed groups by the 9-month (Fig.4E).Steatosis was the major contributor to NAS at the early time point;however,the increase in NAS at late time points was mainly attributed by inflammation,cell death,and fibrosis.The NAS of HFCF-fed groups were 1.09±0.01(1-month),3.26±0.26(3-month),5.20±0.75(5-month),6.50 ± 0.58 (7-month),and 7.63 ± 0.69 (9-month),respectively (Fig.4E).Per NASH CRN criteria,19,20NAS of 1-2 is NAFL,3-4 indicates borderline NASH and 5-8 means definite NASH.Therefore,our results indicate that the HFCF-fed mice sequentially developed steatosis (1 month),borderline steatohepatitis (3 months),definite steatohepatitis (5 months),steatohepatitis with progressive fibrosis (7 months onwards),and spontaneous liver tumor(9 months).Especially,the HFCF-fed mice developed a transition phase of NAFL to NASH at the 3-to 5-month post HFCF diet administration.
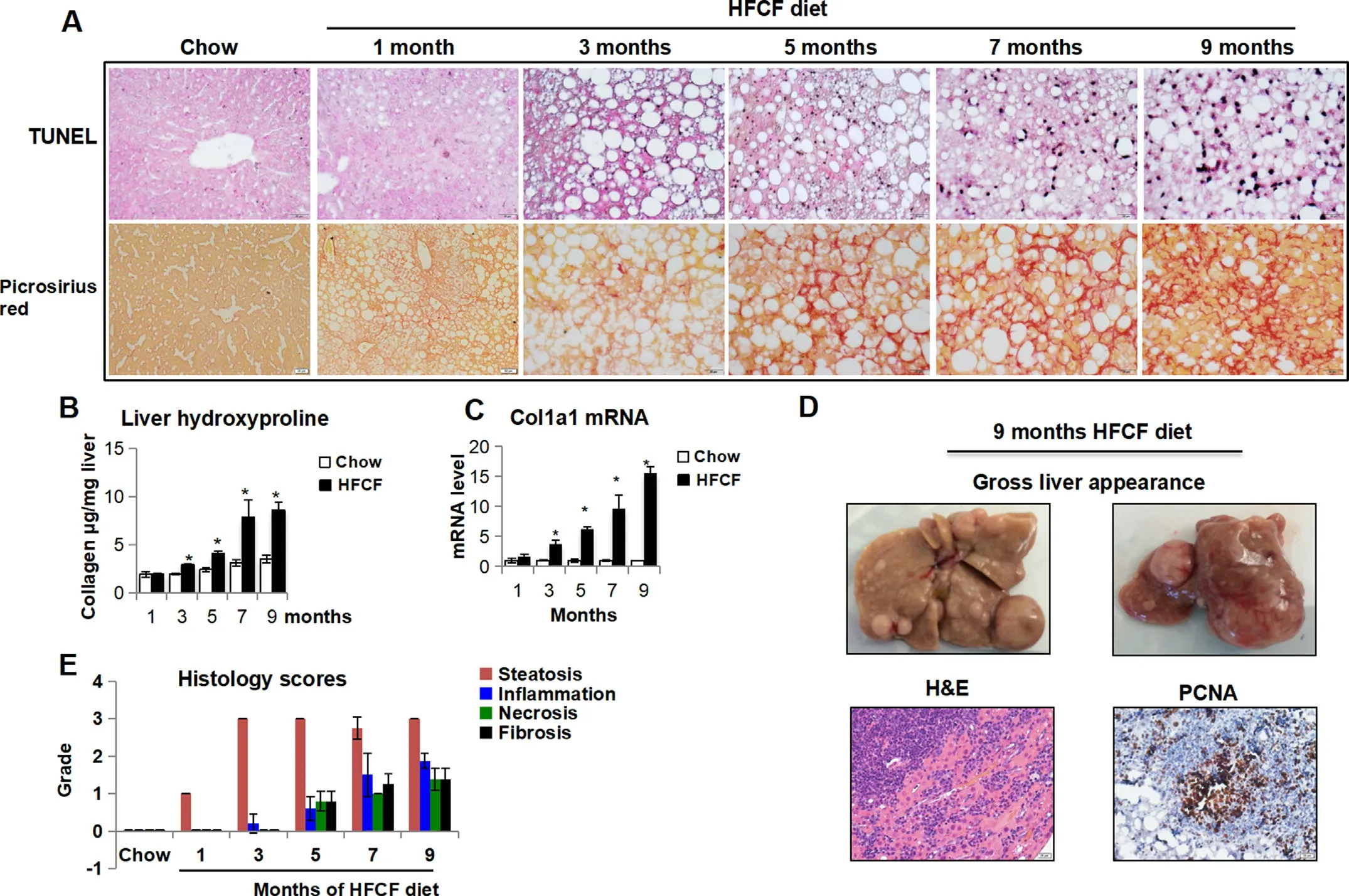
Fig.4.HFCF diet feeding induces hepatocyte cell death,liver fibrosis,and tumor development over time.Two-month-old C57BL/6J male mice were fed chow or an HFCF diet for up to 9 months.(A)Representative images of liver sections stained with TUNEL and Picrosirius Red.Original magnification,×40.(B)Liver collagen content was determined by hydroxyproline assay.(C)The relative mRNA level of gene involved in liver fibrosis was determined by qPCR.(D)Gloss liver appearance and representative images of liver sections stained with hematoxylin-eosin(H&E)or PCNA from mouse on HFCF diet for 9 months.Original magnification,×40.(E)Histology scores of steatosis,inflammation,cell death,and fibrosis.Data presented in Fig.B and C are expressed as the mean ± SEM for 5 mice per group.*P <0.05 versus chow controls.Abbreviations: Col1a1,collagen 1 alpha 1;H&E,hematoxylin and eosin;HFCF,high-fat,cholesterol,and fructose;PCNA,proliferating cell nuclear antigen;qPCR,quantitative polymerase chain reaction;SEM,standard error of the mean;Tnfα,tumor necrosis factor alpha.
3.5.HFCF diet induces remarkable changes to hepatic transcriptome
To pursue an unbiased investigation of early signatures associated with the transition of steatosis to borderline steatohepatitis,we conducted RNA-seq analysis of livers obtained from chow-fed and HFCF-fed mice at 1-month and 3-month time points (data were deposited into GSE135050).The fragments per kilobase of exon model per million read mapped (FPKM) was calculated to quantify the expression levels of genes in four groups,including 1-month chow,1-month HFCF,3-month chow,and 3-month HFCF.The DEGs were determined using a fold change cutoff of 1.50 and FDR <0.05.
Volcano plots demonstrated hepatic transcriptome differences between chow-fed and HFCF-fed groups at 1-month and 3-month time points (Fig.5A).There were 460 genes differentially expressed between the chow-fed and HFCF-fed groups at the 1-month time point.At the 3-month time point,the differences between chow-fed and HFCF-fed groups resulted in 2167 DEGs.Meanwhile,78 genes were differentially expressed in the 1-month chow-fed group compared to the 3-month chow-fed group,and 1869 genes were differentially expressed comparing the 1-month HFCF-fed group to the 3-month HFCF-fed group.A heat map visualization of the hepatic transcriptome with hierarchical clustering was used to determine the expression patterns of different genes in chow-fed and HFCF-fed groups (Fig.5B).The DEGs were clustered into two major types,one with upregulation and the other with downregulation in the HFCF-fed groups compared with the chow-fed groups.The data indicate that HFCF diet feeding induced significant hepatic transcriptomic changes compared to chow-fed groups.Meanwhile,the feeding duration also made a difference,especially for the HFCF-fed groups.
3.6.RNA-seq reveals key gene signatures associated with NAFL transition to borderline NASH in HFCF-fed mice
To reveal the key gene signatures during the transition of NAFL to borderline NASH,we conducted GO enrichment and KEGG pathway analysis with Enrichr.A cutoffP-value of <0.05 was used to select enriched GO terms and KEGG pathways.The GO terms identified a significant enrichment of metabolism genes in the 1-month HFCF group compared to the 1-month chow group.Among them,the top 5 biological processes altered by 1-month HFCF feeding were the regulation of cholesterol metabolism,lipid absorption,acetyl-CoA metabolic process,fatty acid beta-oxidation,and cholesterol absorption(Fig.6A).The steroid biosynthesis,DNA replication,terpenoid backbone biosynthesis,amino acid (glycine,serine,and threonine) metabolism,and PPAR signaling pathway were the top 5 enriched pathways revealed by KEGG (Fig.6A).Fig.6B showed the top 10 deregulated genes between the 1-month chow and HFCF groups.
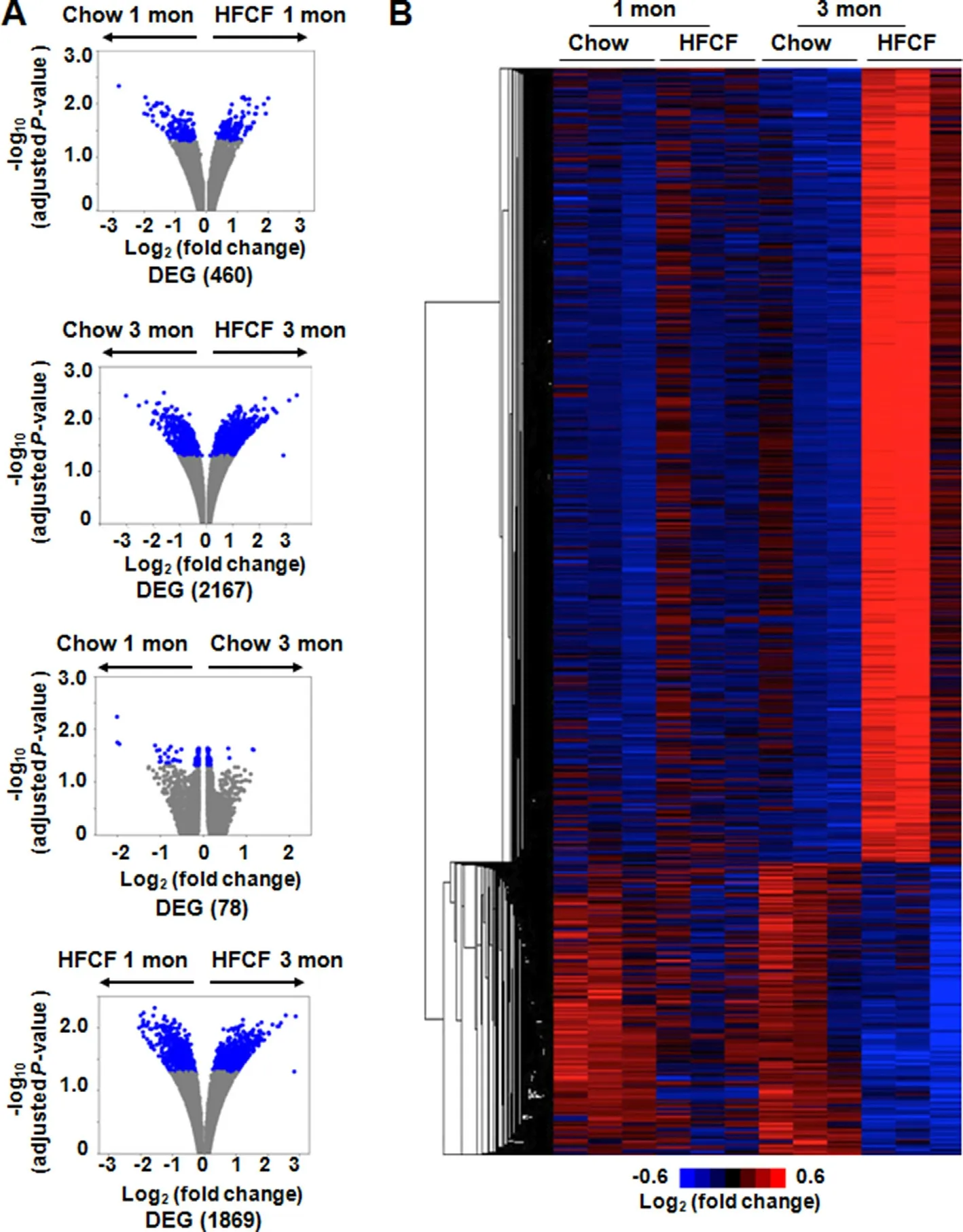
Fig.5.RNA-seq analysis of livers from mice fed chow or HFCF diet for 1 month or 3 months.(A) Volcano plots of mRNAs.The plots were constructed by plotting -log10(adjusted P-value) on the y-axis and log2 (fold change) on the x-axis.Blue blots represent DEGs and grey blots represent mRNAs without significant difference.(B) Hierarchical cluster of representative mRNA expression across biological replicate samples.Red or blue colors indicate high or low gene expression,respectively.Abbreviations: DEGs,differentially expressed genes;HFCF,high-fat,cholesterol,and fructose;RNA-seq,RNA sequencing.
We next continued GO enrichment and KEGG pathway analyses in 3-month feeding groups.Compared to the chow-fed controls,3 months of HFCF feeding significantly altered the expression of genes involved in inflammation and fibrosis.The top 5 biological processes significantly different between the 3-month chow and HFCF groups were ECM organization,leukocyte aggregation,collagen fibril organization,cellular response to bacterial lipopeptide,and citrulline metabolic process(Fig.6C).The top 5 KEGG pathways significantly altered by 3 months of HFCF feeding were arginine biosynthesis,steroid biosynthesis,immune network for IgA production,phagosome,and ECM-receptor interaction.Fig.6D displayed the top 10 deregulated genes between the 3-month chow and HFCF groups.
To identify key gene signatures associated with the progression of NAFL to borderline NASH,DEGs from the 1-month HFCF versus the 3-month HFCF comparison were used for GO enrichment and KEGG pathway analysis.Consistently,genes involved in inflammation and fibrosis were enriched by both analyses.Especially the ECM organization,collagen fibril organization,citrulline metabolic process,T cell migration,and glutathione derivative biosynthetic process were the top 5 biological processes significantly different between the 1-month and the 3-month HFCF-fed groups (Fig.6E).KEGG pathway analysis demonstrated that,compared to 1-month HFCF group,pathways related to arginine biosynthesis,C-type lectin receptor signaling,cytokine-cytokine receptor interaction,PPAR signaling,and glutathione metabolism were significantly altered by 3 months of HFCF diet administration (Fig.6E).The top 10 deregulated genes between 1-month and 3-month HFCF groups were shown in Fig.6F.
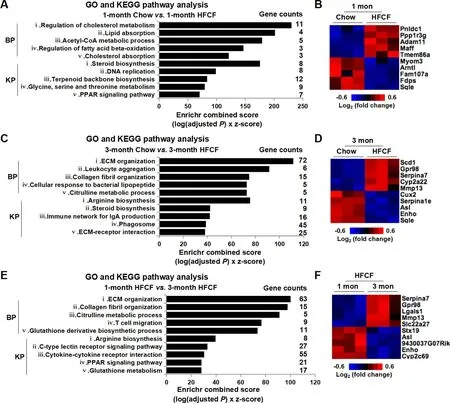
Fig.6.GO enrichment and KEGG pathway analysis of DEGs from mice fed chow or HFCF diet for 1 month or 3 months.(A)The top 5 enriched GO terms and KEGG pathways selected by Enrichr from the comparison of 1-month chow versus 1-month HFCF.(B) Heat map depicting top 10 deregulated genes between 1-month chow and 1-month HFCF groups.(C) The top 5 enriched GO terms and KEGG pathways selected by Enrichr from the comparison of 3-month chow versus 3-month HFCF.(D) Heat map depicting top 10 deregulated genes between 3-month chow and 3-month HFCF groups.(E)The top 5 enriched GO terms and KEGG pathways selected by Enrichr from the comparison of 1-month HFCF versus 3-month HFCF.(F) Heat map depicting the top 10 deregulated genes between 1-month HFCF and 3-month HFCF groups.In A,C,and E,the x-axis represents Enrichr combined score and the y-axis represents GO terms or KEGG pathways.Abbreviations:BP,biological process;DEGs,differentially expressed genes;GO,Gene Ontology;HFCF,highfat,cholesterol,and fructose;KEGG,Kyoto Encyclopedia of Genes and Genomes;KP,KEGG pathway;RNA-seq,RNA sequencing.
3.7.Deregulation of upstream regulators FOXM1 and NELFE in mouse and human NASH
To understand what upstream regulators were responsible for gene signature differences between NAFL and borderline NASH,we performed Encyclopedia of DNA Elements (ENCODE) and ChIP enrichment analysis (ChEA) on DEGs from the comparison of 1-month HFCF versus 3-month HFCF.The use of ENCODE and ChEA allowed us to identify consensus transcription factors.Fig.7A displayed the top 15 consensus transcription factors identified by the ENCODE and ChEA,including Foxm1,estrogen receptor 1 (Esr1),GATA binding protein 1(Gata1),nuclear factor erythroid derived 2-like 2 (Nfe2l2 or Nrf2),GATA binding protein 2 (Gata2),sex determining region Y-box 2 (Sox2),tripartite motif containing 28(Trim28),SMAD family member 4 (Smad4),myogenic differentiation 1 (Myod1),interferon regulatory factor 8 (Irf8),runt related transcription factor 1(Runx1),SUZ12 polycomb repressive complex 2 subunit (Suz12),NF-kB subunit Rela or p65,Nelfe,and tumor protein p63 (Tp63).
To verify whether consensus transcription factors identified during the transition of NAFL to borderline NASH in the mouse model would be relevant to the NAFLD development in humans,we conducted ENCODE and ChEA analyses using DEGs obtained from a human liver microarray dataset (GSE48452).This dataset contains results of microarrays from normal livers and liver samples from different stages of NAFLD,including 14 steatotic livers and 18 NASH livers.The complete list of DEGs between steatotic livers and NASH livers is presented in Supplemental Table 1.Nine consensus transcription factors were revealed by the ENCODE and ChEA analyses(Fig.7B).Strikingly,both FOXM1 and NELFE were identified from the NAFL versus NASH comparison.Fig.7C displayed the heat maps of genes differently regulated by FOXM1 or NELFE in the transition of NAFL to borderline NASH in mouse model.Fig.7D showed fold changes and significantP-values of genes differently regulated by FOXM1 or NELFE in human NAFLD livers.Taken together,our result demonstrated that the upstream regulators FOXM1 and NELFE were deregulated during disease progression from NAFL to NASH.This phenomenon was observed in both mouse NASH and human NASH development.
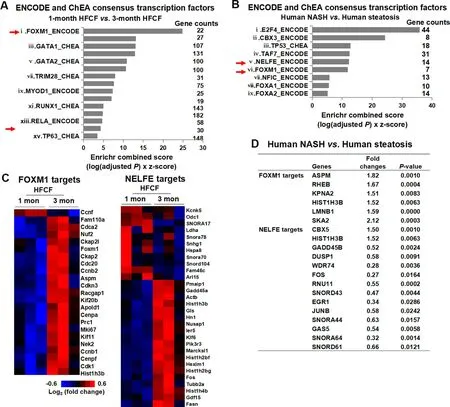
Fig.7.Screen consensus transcription factors by ENCODE and ChEA.(A) The DEGs from the comparison of 1-month HFCF versus 3-month HFCF were submitted to Enrichr.(B)The DEGs from human NASH versus steatosis comparison were downloaded from GSE48452 and submitted to Enrichr.In Fig.A and B,the consensus transcription factors were selected based upon P-value and Enrichr combined score.The x-axis represents Enrichr combined scores,and the y-axis represents consensus transcription factors.(C) Heat map depicting genes regulated by FOXM1 or NELFE in 1-month HFCF versus 3-month HFCF comparison.(D) The fold changes and significant P-values of genes regulated by FOXM1 or NELFE identified from human NASH versus steatosis comparison.Abbreviations: ChEA,ChIP enrichment analysis;DEGs,differentially expressed genes;ENCODE,Encyclopedia of DNA Elements;FOXM1,forkhead box M1;HFCF,high-fat,cholesterol,and fructose;NELFE,negative elongation factor complex member E.
4.Discussion
NAFLD is becoming the most common chronic liver disease worldwide yet the understanding of molecular events that lead to disease progression from simple steatosis to NASH is limited.29Given the clinical importance of NASH,identifying key factors and pathways that promote the disease progression of NAFLD is critically important to develop effective prevention and therapeutic strategies.In the present study,we provided an overview of dynamic changes of NAFLD development in C57BL/6J mice fed a highfat diet enriched in cholesterol and fructose-a model which closely recapitulates the key physiological,metabolic,and histologic changes seen in humans.We demonstrated that upon initiation of HFCF diet,mice became obese and developed dyslipidemia with insulin resistance and sequentially developed simple steatosis,borderline steatohepatitis,steatohepatitis with progressive fibrosis,and eventually liver tumor within the timeframe of 9 months.These stages are consistent with NAFLD progression in humans.To understand the molecular machinery responsible for the transition of simple steatosis to early NASH,we examined hepatic transcriptome profile of chow-fed and HFCF-fed mice at two different feeding periods,1 month and 3 months.We found that the HFCF diet induced remarkable hepatic transcriptomic changes at both time points.The key gene signatures relate to disease progression from NAFL to early NASH in HFCF-fed mice were also found in patients with NAFLD.Therefore,the HFCF-fed mice can serve as a relevant model to identify therapeutic targets and test preventive and therapeutic approaches against NASH.
NAFLD development is strongly associated with obesity.In the current study we found that,upon introduction of HFCF diet,mouse body weight increased rapidly,becoming significantly different from the chow-fed group as early as the 1-month after feeding.However,the increases in liver weight as well as the liver-to-body weight ratio became significant after the 3-month HFCF diet feeding.Similarly,histology showed that lipid accumulation in the liver was severe after the 3-month HFCF diet administration.These data suggested that HFCF-fed mice developed liver steatosis after excessive abdominal fat accumulation.This observation is consistent with the NAFLD development in humans,where studies showed that the prevalence of NAFLD is 58% in overweight individuals but can be as high as 98% in obese individuals.30NAFLD arises from an imbalance between hepatic lipid acquisition(DNL or fatty acids uptake) and removal (fatty acids oxidation or export as VLDL particles),resulting in an increase in lipids flux within hepatocytes that leads to liver steatosis.To understand the regulatory machinery promoting liver steatosis in HFCF-fed mice,we examined the expression of genes responsible for fatty acids uptake,DNL,fatty acids beta-oxidation,and lipid secretion.Pparg,the master transcriptional regulator of adipogenesis and lipid storage,is increased in steatotic livers in both animal models and in NAFLD patients.31,32Disruption of thePparggene in hepatocytes ameliorates steatosis,whereas its overexpression promotes the development of fatty liver through the activation of various lipogenic genes such as fatty acids uptake-related geneCd36and lipid droplet formation-related geneCidec.33,34The HFCF-fed mice displayed sustained upregulation ofPpargand its targets,Cd36andCidec,throughout the 9-month feeding period,suggesting thatPpargregulated increase of fatty acids uptake is an important contributor for liver steatosis.In healthy,lean individuals,DNL contributes to<5%of total triglyceride synthesis in fasting conditions but the rate can be 25% or greater in fed conditions.35BothAcc1andFasnare important for DNL,whereAcc1involves in carboxylation of acetyl-CoA to generate malonyl-CoA andFasncatalyzes malonyl-CoA to produce new saturated fatty acids.Notably,in HFCF-fed mice,bothAcc1andFasnwere decreased at early time points(1 month and 3 months)after diet initiation but increased at later time points(5-9 months) when the liver displayed severe steatosis.The results suggested that DNL is initially suppressed during the onsite of steatosis but significantly promotes severe steatosis once it is activated.Our study is consistent with previous publications which showed that,in NASH patients,DNL rates are increased and could contribute up to 25%of the fatty acids flux into the liver.36-38Given the clinical importance of DNL to NASH development,approaches specifically inhibiting DNL are being tested to treat NASH in humans,including Acc1 inhibitors and liver X receptor (LXR) inverse agonists.39-41
To remove lipids from the liver,lipids can either be secreted as VLDL particles or oxidized through fatty acids oxidation.In HFCFfed mice,bothApoBandMttpwere unchanged during the first 3 months of diet administration but decreased thereafter.This result indicated that the decreased VLDL secretion may exacerbate lipid accumulation,leading to severe steatosis during NASH development.Our observation is supported by previous publications showing that the excessive endoplasmic reticulum stress triggered by hepatic lipids accumulation could decrease VLDL secretion and eventually worsen steatosis and exacerbate NASH progression.42,43CPT1 is an essential enzyme transporting fatty acids from cytosol to mitochondria for beta-oxidation.Studies showed that in NASH patients CPT1 and some other mitochondrial rate-limiting enzymes in beta-oxidation were found reduced by 50%compared to healthy livers.44,45Consistently,we found thatCpt1was transiently increased in the 1-month HFCF-fed group but persistently decreased thereafter.The induction ofCpt1by short-term HFCF feeding could be due toPparaactivation in response to excess uptake of fatty acids in the liver;however,long-term HFCF feeding resulted in augmented malonyl-CoA formation generated from DNL that downregulatesCpt1.46-48
The liver contains a large number of innate and adaptive immune cells.Under physiological conditions a complex network of signals from immune cells and nonimmune cells induce immune tolerance of the liver to antigens.49,50NASH onset and progression involves both innate and adaptive immunity characterized by the activation of resident macrophages and dendritic cells as well as the recruitment of monocytes,neutrophils,and lymphocytes to the liver.51-53These immune cells release cytokines,chemokines,and reactive oxygen species that exacerbate disease progression.Eventually,lobular inflammation is considered the driving force for disease progression in NASH patients.In the present study,introduction of the HFCF diet induced a significantly persistent increase of hepatic macrophages in the liver.Furthermore,the expression of M1 macrophage markerNos2increased while M2 macrophage markersCd163andAgr1both decreased in the livers of HFCF-fed groups.This suggests that pro-inflammatory M1-like macrophages predominated during NAFLD progression while the M2-like phenotype decreased.Besides the increase in macrophages in the liver,the HFCF-feeding also induced a significant increase of dendritic cells,neutrophils,CD4+T cells,and CD8+T cells in the liver with a late increase in B cells,suggesting the involvements of both innate and adaptive immunity in NASH pathogenesis.Our finding is important as the vast body of existing literature emphasize the critical role of innate immune mechanisms as key elements in promoting hepatic inflammation during NASH pathogenesis.Our observations indicate that while macrophages indeed represent the most persistently activated cells in NAFLD development,other immune cells such as T and B lymphocytes involved in adaptive immunity also participate in disease progression.Indeed,increasing evidence points to the role of adaptive immunity as an additional factor amplifying hepatic inflammation during NASH progression.53,54
The molecular nature of hepatic transcriptome profile during NAFL transition to NASH,especially the early phase of NASH,is incompletely understood.By using RNA-seq,we observed a strong induction of key signatures related to ECM organization and immune responses such as leukocyte aggregation,chemotaxis,phagocytosis,and dendritic cell differentiation during the NAFL progression to early NASH.Overall these results are consistent with previous hepatic transcriptomic studies in NASH mouse models and patients.17,55,56One striking finding of this study is that the expression of genes regulated by transcription factorFoxm1 and Nelfewere significantly different between NAFL and NASH,which was observed in both NAFLD mouse model and NAFLD patients.FOXM1 is a proliferation-associated transcription factor expressed during cell cycle.57FOXM1 controls the critical expression of genes involved in cancer initiation,cancer progression,cancer metabolism,and drug resistance.58,59Increase in hepatic FOXM1 is reported in human HCC and inhibition of FOXM1 by targeting mevalonate pathway is beneficial for HCC treatment.60,61Additionally,a recent study demonstrates that insulin signaling promotes adaptive pancreatic β cell proliferation through FOXM1/polo-like kinase 1 (PLK1)/centromere protein A (CENP-A)pathway that is an essential component to delay or prevent progression of diabetes,62suggesting a potential role of FOXM1 in metabolic disease.The role of FOXM1 in NAFLD development especially in NAFL transition to NASH remains currently unknown and is an important unsolved question.NELFE is originally identified as part of a complex termed negative elongation factor(NELF)repressing RNA polymerase II transcript elongation,but a recent study reveals NELFE as an RNA-binding protein.63Although the molecular function of NELFE in liver metabolism is largely unknown,high levels of NELFE have been demonstrated in HCC.64Furthermore,NELFE is reported as an oncogenic protein in HCC through the regulation of MYC signaling.63Whether NELFEregulated pathways could contribute to the onset and progression of NAFLD remains unknown and worthy of further investigation.
Our study provides a unique overview of the dynamic changes in NAFLD mouse model that is consistent with disease progression in humans.However,we acknowledge that there are several limitations in the current study.The cholesterol concentration in the HFCF diet is 2%,which is higher than 0.2% cholesterol in most Western diet.Indeed,in both models the dietary cholesterol and fat synergize to trigger liver injury.65,66In the present study monocytederived macrophages in HFCF-fed livers could not be distinguished from resident Kupffer cells by F4/80 staining.Recent studies employed novel tools such as single-cell RNA sequencing have demonstrated that resident Kupffer cells diminished during NAFLD development,and the marked hepatic influx of monocytes and monocyte-derived macrophages contribute to the uncontrolled inflammatory environment that dampens liver injury and exacerbates NASH progression.67,68Therefore,understanding mechanisms by which resident Kupffer cells and infiltrated monocytederived macrophages differently contribute to NASH pathogenesis is an important topic and requires further investigation.Moreover,several questions such as the changes of hepatic transcriptome profile and signaling pathway in the late stages of NAFLD and the functional impacts of FOXM1 and NELFE on NAFLD development also remain to be resolved.We look forward to future scientific endeavors to answer these important questions.
Authors’ contributions
N.Magee,F.Ahamed,N.Eppler,E.Jones,P.Ghosh,L.He,and Y.Zhang performed experiments and analyzed data.Y.Zhang conceived and supervised the study and wrote manuscript.All authors have reviewed the results and approved the final version of manuscript.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Acknowledgements
The authors would like to thank Dr.Maura O'Neil for evaluating the histologic findings.This work was supported by the National Institutes of Health grants R01DK119131,K22CA184146,P20 GM103549,P30GM118247,P20GM103418,T32ES007079,UL1 TR002366 and KUMC Enhancement Award,American Association for the Study of Liver Diseases (AASLD) Bridging Award,and American Cancer Society (ACS) Institutional Research Grant (IRG)16-194-07 to Y.Zhang.The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.livres.2022.11.001.
杂志排行

Liver Research的其它文章
- Pathogenesis of fatty liver diseases and hepatocellular carcinoma☆
- Prevalence,diagnosis,treatment,and associated factors of hepatitis C in the United States from 1999 to 2018: A population-based crosssectional study☆
- Effects of apical sodium-bile acid transporter inhibitor and obeticholic acid co-treatment in experimental non-alcoholic steatohepatitis☆
- Prediction of effective percutaneous transhepatic biliary drainage in patients with hepatocellular carcinoma: A multi-central retrospective study☆
- MUTYH is a potential prognostic biomarker and correlates with immune infiltrates in hepatocellular carcinoma☆
- Chemical basis of pregnane X receptor activators in the herbal supplement Gancao (licorice)☆