Mdm2过表达诱导ALK阳性非小细胞肺癌克唑替尼耐药的实验研究
2023-01-03刘嘉欣李姗姗吕镗烽叶明翔
刘嘉欣,李姗姗,王 栋,吕镗烽,印 洁,宋 勇,叶明翔
210002 南京,东部战区总医院呼吸与危重症医学科
2007年日本科学家Hiroyuki Mano从1例晚期肺腺癌患者肿瘤组织中克隆出EML4-ALK融合基因,该融合基因包含了间变性淋巴瘤激酶(anaplastic lymphoma kinase,ALK)结构域,可持续激活PI3K/Akt信号通路,具有使细胞恶变的能力[1]。除了EML4-ALK,从肺癌组织中还鉴定出KIF5B-ALK、TFG-ALK、NPM3-ALK等融合形式,但不论是何种融合类型,这些肺癌组织都高表达ALK且ALK蛋白发生自动磷酸化(auto-phosphorylation),使用特异性抗体可检出ALK融合蛋白[2-3]。3%~5% 非小细胞肺癌(non-small cell lung cancer,NSCLC)发生ALK基因融合,称为ALK阳性NSCLC,这部分患者对ALK靶向治疗药物(例如克唑替尼,阿来替尼,布加替尼等)敏感。研究表明克唑替尼(Crizotinib)一线治疗ALK阳性NSCLC的有效率达74%,显著优于含铂两药方案静脉化疗(45%),疾病无进展期(progression-free survival,PFS)明显延长(10.9个月vs.7.0个月),克唑替尼组患者1年生存率高达84%[4]。因此ALK靶向药物推荐用于ALK阳性NSCLC标准一线治疗。
不幸的是大部分ALK阳性NSCLC患者在克唑替尼靶向治疗10个月后产生获得性耐药(acquired resistance),相关耐药机制包括ALK激酶结构域点突变(C1156Y,L1196M),ALK扩增,EGFR和c-kit旁路活化等[5-6],前者可以通过升级ALK靶向药物(色瑞替尼,阿来替尼,劳拉替尼)进而克服耐药,联合EGFR和c-kit靶向药物则可逆转旁路途径引起的耐药。然而,仍有30%患者耐药机制尚未明确,这些患者缺少有效的治疗方案,生活质量迅速恶化,预后较差。故探究未知的ALK靶向耐药机制一直是基础研究和临床实践的热点和难点。
鼠双微体基因2(murine double minute 2,Mdm2)定位于12q13-14,编码分子量90kD的Mdm2蛋白。Mdm2蛋白主要在胞浆和细胞核内表达,通过降解p53等细胞周期蛋白促进细胞增殖[7]。骨肉瘤、软组织肉瘤、卵巢癌、淋巴瘤、胃癌、NSCLC等实体瘤中通常可以检出Mdm2高表达,并且Mdm2表达水平和肿瘤侵袭转移能力正相关,Mdm2高表达的肿瘤患者生存期短,提示Mdm2是驱动肿瘤细胞恶性表型的重要因子,目前亦开发出处于临床研究阶段的Mdm2抑制剂Nutlin-3,与化疗联合用于治疗晚期恶性肿瘤患者[8-9]。然而Mdm2对ALK靶向治疗敏感性的调控作用仍不甚清楚,Mdm2过表达能否导致耐药表型亟待明确。因此,本研究拟使用MTT、平板克隆、TUNEL染色等多种实验方法,探究Mdm2 过表达在H3122细胞中对ALK靶向治疗敏感性的影响,采用Western blot实验初步探究其导致耐药的分子机制,以期为ALK靶向治疗耐药后药物的研发提供新的靶点。
1 材料与方法
1.1 主要试剂
RPMI 1640、胎牛血清购自Gibco 公司,Lipofectamine 3000转染试剂购自Invitrogen公司,克唑替尼购自Selleck公司,MTT、DMSO购自Sigma 公司,吉姆萨染液购自碧云天公司,TUNEL试剂盒购自诺唯赞公司, p-ALK、ALK、p-Akt、Akt、pErk、Erk、E-cadherin、Vimentin、Snail、Slug抗体购自Cell Signaling Technology 公司,Mdm2抗体购自Santa Cruz公司, GAPDH 抗体购自Proteintech 公司。
1.2 细胞培养和转染
ALK阳性NSCLC H3122细胞由麻省总医院胸部肿瘤中心Jeffrey Engelman教授惠赠,用含10% 胎牛血清的RPMI 1640培养基置于37℃、5% CO2培养箱中常规培养。
转染前1天,将H3122细胞接种到6 cm 培养皿中,次日使用Lipofectamine 3000 转染空载体(empty vector,EV)或Mdm2过表达质粒,转染48 h后用于功能学实验。
1.3 MTT实验
转染后,将空载或Mdm2 过表达的H3122细胞以3 000/孔密度接种到96孔板中,次日加入梯度克唑替尼(0、1、10、100、1 000 nmol/L),继续培养48 h。培养结束后向各孔加入5 mg/mL MTT溶液,孵育4 h后吸弃上清,DMSO溶解甲瓒结晶,酶标仪读取各孔490 nm处吸光度值[D(490)]。
1.4 平板克隆实验
转染后,将空载或Mdm2 过表达的H3122细胞以1 000/孔密度接种到3.5 cm培养皿中,次日待细胞贴壁后加入终浓度为20 nmol/L 克唑替尼,每3天换液一次,置于37 ℃、5% CO2培养箱中培养。培养约2周有肉眼可见细胞克隆时停止培养,甲醇—冰醋酸固定细胞克隆,1% 吉姆萨染液对细胞克隆进行染色。
1.5 TUNEL细胞凋亡检测
常规制备细胞爬片,4%多聚甲醛室温固定30 min,Proteinase K溶液室温孵育5 min,每个样本滴加100 μL 1×Equilibration Buffer室温孵育30 min,50 μL TdT孵育缓冲液室温孵育1 h,PBS漂洗后DAPI室温染色5 min。在荧光显微镜下分析各组细胞爬片,620 nm波长下检测Alexa Fluor 640红色荧光,460 nm波长下检测DAPI蓝色荧光。
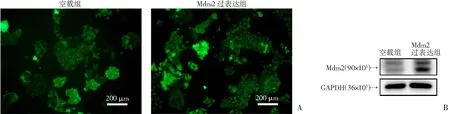
A:荧光显微镜下检测质粒转染效率;B:Western blot检测转染后Mdm2蛋白表达情况
1.6 Western blot实验
细胞经过药物处理后使用RIPA裂解液冰上裂解,BCA法检测细胞蛋白样品浓度,SDS-PAGE凝胶电泳后湿法转膜,5% 脱脂牛奶室温封闭1 h。一抗4 ℃冰箱过夜孵育后HRP标记的二抗室温孵育1 h,TBST漂洗后进行ECL化学发光显像。
1.7 统计学分析
每组实验至少重复3次,结果采用SPSS 25.0进行统计分析,GraphPad Prism 8.0绘制统计图。两组比较采用Student’st检验,以P<0.05视为差异具有统计学意义。
2 结果
2.1 过表达Mdm2诱导H3122细胞克唑替尼耐药
H3122细胞为EML4-ALK阳性NSCLC,该细胞对ALK靶向治疗敏感,故本研究主要在H3122细胞中评估Mdm2表达状态与克唑替尼敏感性的关系。在pEGFP-N3表达载体中插入Mdm2 cDNA序列,构建了Mdm2表达载体,该载体在N端有EGFP序列,可在荧光显微镜下观察目的基因表达情况。H3122细胞转染pEGFP-N3-Mdm2编码质粒或pEGFP-N3空载体(EV)质粒后,荧光显微镜下可见明亮的绿色荧光,提示质粒转染成功(图1A),将空载组或Mdm2过表达组的细胞提取蛋白后,经Western blot验证,发现Mdm2过表达组H3122细胞Mdm2蛋白表达量明显增加,说明转染质粒在细胞内表达(图1B)。
通过MTT实验评估Mdm2表达状态对克唑替尼敏感性的影响。结果显示:克唑替尼能有效抑制H3122 空载组细胞增殖,药物处理48 h估算IC50值约189.5 nmol/L;相反,H3122 Mdm2过表达组细胞对克唑替尼处理不敏感,细胞可以在低浓度克唑替尼条件下继续增殖,估算克唑替尼 IC50值约674.3 nmol/L。加入不同浓度的克唑替尼后,Mdm2 过表达组的细胞存活率均高于空载组细胞,差异均具有统计学意义(P<0.05,图2)。因此,过表达Mdm2可以诱导H3122细胞对克唑替尼的耐药性。
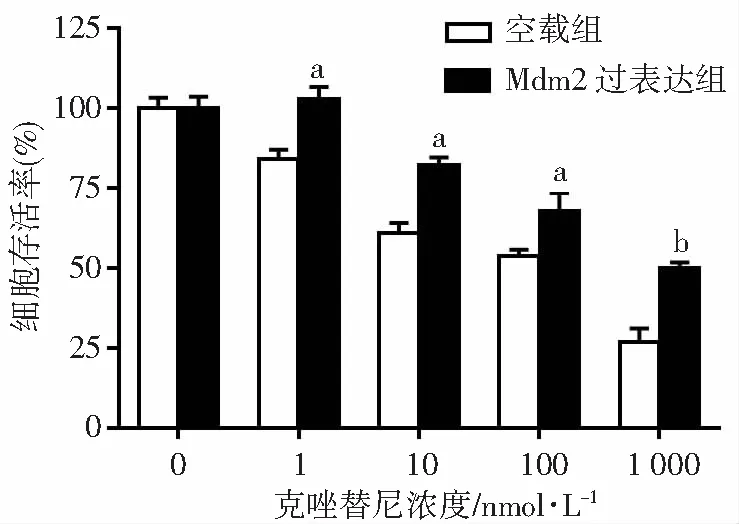
a:P<0.05,b:P<0.01,与空载组比较
2.2 Mdm2促进H3122细胞克隆性增殖
MTT实验仅能反应药物对肿瘤细胞的短期杀伤作用,为了评估Mdm2诱导克唑替尼耐药性的作用是否持久稳定,将H3122 空载组细胞和H3122 Mdm2过表达组细胞接种培养皿后在低浓度克唑替尼(20 nmol/L)刺激下长时间培养,进行平板克隆实验。本研究发现DMSO溶剂组H3122 空载组细胞和H3122 Mdm2过表达组细胞均出现克隆性增殖,培养皿中可见大量细胞克隆(图3);经克唑替尼处理后H3122 空载组细胞的增殖被显著抑制,培养皿中很少形成细胞克隆,而H3122 Mdm2过表达组细胞在克唑替尼存在条件下仍能继续生长,培养2周后仍有较多细胞克隆形成,说明Mdm2过表达可以稳定持久地诱导H3122细胞ALK靶向治疗耐药表型。
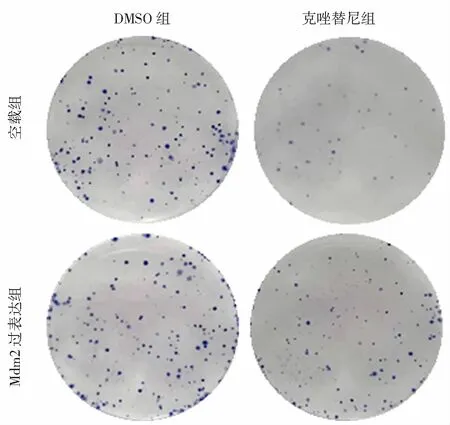
图3 平板克隆实验评估Mdm2过表达对H3122细胞克
2.3 过表达Mdm2导致H3122细胞凋亡抵抗
本研究探究了Mdm2诱导的H3122细胞克唑替尼耐药是否与凋亡减少有关。细胞凋亡过程通常伴随DNA断裂,使用荧光素标记的dUTP在脱氧核糖核苷酸末端转移酶(terminal deoxynucleotidyl transferase, TdT)催化下可连接到凋亡细胞断裂DNA 的3’-OH末端,原位显示发生凋亡的细胞。本研究发现溶剂DMSO处理组H3122 空载组和H3122 Mdm2过表达组细胞几乎不发生凋亡,视野下几乎看不到提示凋亡的红色荧光(图4);1 μmol/L 克唑替尼处理6 h后H3122 空载组细胞视野下可见大片红色荧光,说明克唑替尼诱导了细胞凋亡,而H3122 Mdm2过表达组细胞经克唑替尼处理后并未发生凋亡。故过表达Mdm2可以导致H3122细胞对克唑替尼耐受,并且对抗克唑替尼诱导的细胞凋亡。

图4 TUNEL染色实验检测克唑替尼诱导H3122 空载组细胞和H3122 Mdm2过表达组细胞凋亡情况
2.4 过表达Mdm2诱导H3122细胞克唑替尼耐药的机制探究
上述结果证实过表达Mdm2可以诱导ALK靶向治疗耐药,那么导致耐药的具体机制如何?本研究首先探究了过表达Mdm2是否对PI3K/Akt和MAPK/Erk信号活化状态造成影响。本研究发现克唑替尼可以有效抑制对照组H3122 空载细胞ALK磷酸化,随着pALK表达量减少,PI3K/Akt和MAPK/Erk信号通路也被阻断,Akt和Erk磷酸化水平被完全抑制(图5A);Mdm2过表达后PI3K/Akt和MAPK/Erk信号仍然能被克唑替尼抑制,如图5A所示,克唑替尼能够完全抑制H3122 Mdm2过表达组细胞ALK、Akt和Erk磷酸化水平,说明Mdm2并不是通过激活PI3K/Akt和MAPK/Erk信号导致H3122细胞对克唑替尼耐药。
本研究发现过表达Mdm2后H3122细胞形态上出现改变:H3122 空载组细胞大多呈圆形,细胞间缝隙小,细胞黏附力强,细胞聚集成团、成片生长;H3122 Mdm2过表达组细胞形态上呈扁圆形或梭形,细胞表面有伪足突起样结构,细胞间粘附不再紧密,细胞间有缝隙(图5B)。上述形态学特征让我们联想到与肿瘤侵袭转移密切相关的上皮间质转化(epithelial mesenchymal transition,EMT)。
因此,通过Western blot检测了H3122 空载组细胞和H3122 Mdm2过表达组细胞EMT标记物的表达情况,发现H3122 空载组细胞主要表达上皮细胞标记物E-钙粘素 (E-cadherin),而转录因子Snail和Slug处于低表达状态,H3122 Mdm2过表达组细胞主要表达间质细胞标记物波形蛋白 (Vimentin),Snail和Slug表达量明显增加(图5C)。因此,过表达Mdm2可以诱导H3122细胞EMT过程的产生,后者可能促进克唑替尼耐药和凋亡抵抗。
3 讨论
越来越多的ALK阳性NSCLC患者从靶向治疗中获益,患者的生活质量和生存时间得到了极大提高,且随着二代和三代ALK靶向药物的问世,患者的获益得到进一步提高,实现长时间带瘤生存已成为现实[10-11]。但是ALK靶向治疗的获得性耐药不可避免,耐药机制尚未完全解析。本研究发现Mdm2过表达可以诱导H3122细胞克唑替尼耐药表型,并且H3122 Mdm2过表达组细胞抵抗克唑替尼诱导的凋亡,过表达Mdm2后细胞发生EMT,因此认为Mdm2有可能通过改变细胞上皮细胞表型促进克唑替尼耐药。
既往对Mdm2的研究主要关注它在细胞周期调控方面的作用:Mdm2通过降解p53,减少p21、p16INK4a、p14ARF等下游细胞周期相关蛋白的表达,增加细胞周期蛋白cyclin E活性,最终加速细胞周期转化,促进细胞分裂[12]。真实世界研究中Mdm2基因扩增在NSCLC发生率约21%,与年龄、性别、疾病分期和组织学分级等临床病理特征无明确相关性,Mdm2扩增组患者中位无进展生存期(progression-free survival,PFS) 3个月,中位总生存期(overall survival,OS)9个月,而非扩增组中位PFS长达31个月,中位OS 33个月,说明Mdm2表达水平越高,患者预后越差[13]。体外研究还发现Mdm2高表达影响肿瘤细胞对烷化剂、拓扑异构酶抑制剂等化疗药物的敏感性,可能机制为Mdm2与胞内Topo Ⅱ相结合下调其含量,使肿瘤细胞对化疗药敏感性下降,使用RNAi降低胞内Mdm2表达可稳定Topo Ⅱ表达,缓解化疗药物耐受[14]。上述结果均提示Mdm2参与了肿瘤细胞分裂、增殖、耐药等恶性生物学过程,因此靶向Mdm2可能作为肿瘤治疗的新思路。目前在研的Mdm2抑制剂有Nutlin-3、RG7112、MI-77301、MI-888等,这些小分子抑制剂阻断Mdm2-p53相互作用从而阻滞细胞周期,诱导细胞凋亡,在肿瘤动物模型中甚至实现了肿瘤的完全消退,在人体内初步药代动力学和药理活性研究亦显示较好的抗肿瘤活性,主要不良反应包括中性粒细胞减少和腹泻,后续可能通过优化药物剂量来缓解[15-16]。本研究重点关注了Mdm2表达状态与ALK靶向治疗敏感性之间的联系,发现Mdm2过表达可导致H3122细胞克唑替尼耐药,这与目前已知的ALK靶向治疗耐药机制不尽相同:①Mdm2不直接引起ALK磷酸化和ALK总蛋白表达水平变化,且H3122 Mdm2过表达组细胞的ALK磷酸化水平能够有效地被克唑替尼抑制,说明Mdm2并不通过影响ALK信号通路诱导耐药;②Mdm2没有激酶活性,不能够像EGFR、c-kit等激酶那样旁路激活下游PI3K/Akt和MAPK/Erk信号通路,在Western blot实验中也发现Mdm2过表达不能维持Akt和Erk磷酸化。因此,Mdm2过表达是诱导克唑替尼耐药的新机制。

A:H3122 空载组细胞和H3122 Mdm2过表达组细胞经梯度克唑替尼(0、200、500、1 000 nmol/L)处理6 h,Western blot检测ALK及下游信号通路;B:H3122 空载组细胞和H3122 Mdm2过表达组细胞形态学特点;C:Western blot检测H3122 空载组细胞和H3122 Mdm2过表达组细胞EMT标记物和转录因子表达情况
因此,本研究对Mdm2诱导的耐药机制进行了初步探究,特别是H3122 Mdm2过表达组细胞在形态学上的改变引起了我们的注意,通过形态学对比和Western blot,本研究认为Mdm2过表达的Crizotinib耐药细胞发生了EMT。EMT不仅是细胞形态学上的改变,还伴有细胞骨架重塑、细胞运动能力增强、细胞极性改变等,细胞发生EMT后细胞间粘附减弱,细胞运动能力增强,且表现出肿瘤干细胞特征,对抗肿瘤治疗敏感性下降。而在分子水平,其发生过程是上皮细胞标记E-cadherin表达减少,而间质细胞标记Vimentin表达增加,细胞间粘附变弱,运动能力增强,逐步浸润周围组织,并导致远处转移。在这个过程中Snail、Slug等转录因子活性增强,与E-cadherin上游启动子结合后抑制其转录[17-18]。在研究中,H3122 Mdm2过表达组细胞Snail和Slug等调控EMT的转录因子表达水平增加,使E-cadherin转录受到抑制,细胞获得间质表型,进而对克唑替尼敏感性下降,出现凋亡抵抗,最终导致克唑替尼耐药。因此,在靶向治疗前评估Mdm2表达水平有可能预测患者靶向治疗效果。
综上所述,本研究发现Mdm2过表达有可能通过诱导细胞EMT促进克唑替尼耐药,这种全新的耐药机制提示临床上通过可以检测基线状态下Mdm2表达水平预测患者后续出现耐药的风险,联合Mdm2抑制剂有可能逆转ALK靶向治疗耐药。
