lmmunological classification of hepatitis B virus-positive hepatocellular carcinoma by transcriptome analysis
2023-01-03ShengWeiLiLiFanHanYinHeXiaoShengWang
Sheng-Wei Li, Li-Fan Han, Yin He, Xiao-Sheng Wang
Abstract BACKGROUND Hepatitis B virus (HBV) infection is a major factor responsible for HBV+ hepatocellular carcinoma (HCC).AIM An immunological classification of HBV+ HCC may provide both biological insights and clinical implications for this disease.METHODS Based on the enrichment of 23 immune signatures, we identified two immunespecific subtypes (Imm-H and Imm-L) of HBV+ HCC by unsupervised clustering.We showed that this subtyping method was reproducible and predictable by analyzing three different datasets.RESULTS Compared to Imm-L, Imm-H displayed stronger immunity, more stromal components, lower tumor purity, lower stemness and intratumor heterogeneity, lower-level copy number alterations, higher global methylation level, and better overall and disease-free survival prognosis.Besides immune-related pathways, stromal pathways (ECM receptor interaction, focal adhesion, and regulation of actin cytoskeleton) and neuro-related pathways (neuroactive ligand-receptor interaction, and prion diseases) were more highly enriched in Imm-H than in Imm-L.We identified nine proteins differentially expressed between Imm-H and Imm-L, of which MYH11, PDCD4, Dvl3, and Syk were upregulated in Imm-H, while PCNA, Acetyl-a-Tubulin-Lys40, ER-α_pS118, Cyclin E2, and β-Catenin were upregulated in Imm-L.CONCLUSION Our data suggest that “hot” tumors have a better prognosis than “cold” tumors in HBV+ HCC and that “hot” tumors respond better to immunotherapy.
Key Words: Hepatitis B virus; Hepatocellular carcinoma; Immunological classification; Transcriptomics; Tumor immunity; Cancer immunotherapy
lNTRODUCTlON
Hepatocellular carcinoma (HCC) is a major cancer of the liver that constitutes around 90% of liver cancer cases[1].Although traditional therapeutic approaches, including surgery, chemotherapy, radiotherapy, and targeted therapy, are effective in improving the survival of HCC patients, the overall survival prognosis of HCC patients is generally unfavorable[2].More recently, immunotherapy, such as immune checkpoint blockade (ICB), has shown success in the treatment of various cancers, including HCC[3].However, only a small proportion of cancer patients respond well to immunotherapies to date[4].To this end, certain predictive markers for cancer immunotherapy responses have been uncovered,e.g., PD-L1 expression[5], tumor mutation burden (TMB)[6], and mismatch repair deficiency[7].In addition, the tumor immune microenvironment (TIME) plays an important role in immunotherapy responses[8].Overall, the “hot” tumors infiltrated by a substantial number of tumor-infiltrating lymphocytes (TILs) are more responsive to immunotherapies, compared to the “cold” tumors lacking TILs[9].Hence, an investigation of the TIME in HCC would aid in the prediction of immunotherapy responses.
With the recent emergence of large-scale cancer genomics data, such as the Cancer Genome Atlas (TCGA) (https://portal.gdc.cancer.gov/) and International Cancer Genome Consortium (ICGC) (https://dcc.icgc.org/), many studies have investigated the TIME in HCC based on these data[10-12].For example, Gaoet al[10] identified four immune-relevant subtypes of HCC based on the enrichment of 13 signatures and revealed significantly different molecular and clinical characteristics among these subtypes.Siaet al[11] uncovered an immune subclass of HCC representing nearly 25% of HCC cases, based on gene expression profiles in tumor, stromal, and immune cells.Based on the enrichment of immune cell subpopulations, Farhaet al[12] identified two immune clusters of HCC, and found that the cluster enriched with M0 macrophages had a worse prognosis.
Despite these previous studies[10-12], the discovery of immune-specific subtypes of hepatitis B viruspositive (HBV+) HCC is worth investigating, considering that HBV infection is a major cause of HCC[13].In this study, to characterize the immunological landscape of HBV+ HCC, we identified its immune-specific subtypes by the unsupervised machine learning in transcriptomic data.Furthermore, we comprehensively compared the clinical and molecular features of these subtypes.Our analysis would provide new insights into the HBV+ HCC immunity and its associated clinical and molecular features, as well as potential clinical implications for the management of this disease.
MATERlALS AND METHODS
Datasets
We obtained the TCGA Hepatocellular Carcinoma (TCGA-LIHC) dataset, including transcriptomes (RSEM-normalized RNA-Seq gene expression profiles), somatic mutations (“maf” file), somatic copy number alterations (SCNAs) (“SNP6” files), normalized protein expression profiles by Reverse Phase Protein Array (RPPA), and clinical data, from the Genomic Data Commons (GDC) Data Portal (https://portal.gdc.cancer.gov/).We obtained other two HCC transcriptomic datasets (GSE14520 and GSE121248) from the Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/).A description of these datasets is provided in Supplementary Table 1.
Single-sample gene set enrichment analysis
We evaluated the enrichment of an immune signature or pathway in a tumor by the single-sample geneset enrichment analysis (ssGSEA)[14].This method extends the GSEA method[15] to obtain the enrichment scores of input gene sets in specimens with input of gene expression matrices and marker or pathway gene sets.The marker or pathway gene sets of immune signatures or pathways are presented in Supplementary Table 2.
Identification of immune-specific subtypes of HBV+ HCC
By hierarchical clustering, we identified immune-specific subtypes of HBV+ HCC based on the enrichment scores of 23 immune signatures.The 23 immune cell types included Pro-inflammatory cytokines, APC co-inhibition, APC co-stimulation, Cytolytic activity, Immune cell infiltrate, Inflammation-promoting, Interferon, M1 macrophage, MHC Class I, Myeloid-derived suppressor cell, T cell co-inhibition, T cell exhaustion, Th1 cell, Th2 cell, TILs, Activated dendritic cell, Eosinophil, Immature dendritic cell, Macrophage, Monocyte, Natural killer cell, Plasmacytoid dendritic cell, Activated B cell.Before clustering, we performed the Z-score normalization of the ssGSEA scores and converted them into distance matrices using the R function “dist” with the following parameter: Method = “euclidean.” We performed the hierarchical clustering using the function “hclust()” in the R package“Stats” with the following parameters: method = “ward.D2” and members = NULL.
Class prediction
We conducted classification with the random forest (RF) algorithm.In the RF, the size of trees was 500, and the features were the 23 immune signatures.The prediction performance, namely the accuracy and weighted F-score, were reported.We performed this procedure using the R package "randomForest".
Survival analysis
We compared overall survival (OS) and disease-free survival (DFS) rates between two classes of samples with the Kaplan-Meier (K-M) method[16].K-M curves were used to show the differences in survival rates, and log-rank tests were utilized to evaluate their significance.
Calculation of TMB, SCNAs, Stemness scores, intratumor heterogeneity scores, immune scores, and tumor purity in tumors
A tumor’s TMB was defined as its total count of somatic mutations.We used GISTIC2[17] to calculate SCNA frequencies and amplitudes in the immune-specific subtypes of HBV+ HCC with the input of “SNP6” files.A tumor’s stemness score was its ssGSEA score of the stemness marker genes, as shown in Supplementary Table 1.We measured intratumor heterogeneity (ITH) levels with the DEPTH algorithm[18], which evaluates ITH levels based on gene expression profiles.We assessed immune scores and tumor purity of bulk tumors using ESTIMATE[19].The immune scores indicate the levels of tumor immune infiltration, and tumor purity represents the fraction of tumor cells within a tumor bulk.
Pathway and gene ontology enrichment analysis
To identify pathways that are more enriched in one group compared to another group, we first uncovered the genes significantly upregulated in the group versus another group by Student'sttests using thresholds of false discovery rate (FDR) < 0.05 and mean gene expression levels’ fold change (FC) > 2.We then input the upregulated genes into the GSEA web tool[15] to obtain the Kyoto Encyclopedia of Genes and Genomes (KEGG)[20] pathways using a threshold of FDR < 0.05, which were more enriched in that group versus another class.Besides, we used the weighted gene co-expression network analysis (WGCNA)[21] to identify the gene modules of co-expressed genes.Based on the expression correlations between the hub genes in gene modules, we identified the gene ontology (GO) terms showing significant correlations with specific traits.The WGCNA analysis was performed with the R package “WGCNA” (version 1.68).
Statistical analysis
In comparisons of two classes of normally distributed data, including gene expression levels, protein expression levels, and the ratios of immune-stimulatory to immune-inhibitory signatures, we used twotailed Student’sttests.In comparisons of two classes of data that were not normally distributed, including immune scores, stemness scores, ITH scores, TMB, and global methylation levels, we performed one-tailed Mann-WhitneyUtests.We utilized the Spearman method to calculate correlations between immune scores and protein expression levels or pathways’ enrichment scores, and reported correlation coefficients (ρ) andPvalues.We employed the Benjamini-Hochberg method[22] to calculate FDR for adjusting for multiple tests.We performed all statistical analyses in the R programming environment (version 4.1.3).
RESULTS
Clustering analysis identifies two immune-specific subtypes of HBV+ HCC
This analysis identified two subtypes of HBV+ HCC according to the enrichment scores of 23 immune signatures by hierarchical clustering, consistently in three transcriptomic datasets (TCGA-LIHC, GSE14520, and GSE121248) (Figure 1A).We termed the subtypes Imm-H and Imm-L, respectively, which showed high and low immune signature enrichment, respectively (Figure 1A).To explore whether this classification is predictable, we took one of the three datasets as the training set and the rest as test sets, in turn, to predict the subtypes by RF based on the attribute values (ssGSEA scores of the 23 immune signatures).The 10-fold CV accuracies and weighted F-scores in the training sets were all above 90%.The prediction accuracies and weighted F-scores in test sets with TCGA-LIHC or GSE121248 as the training set were not less than 90% (Figure 1B).These results demonstrate that the subtyping is predictable.
We further compared the expression levels of 25 genes encoding human leukocyte antigens between the subtypes.Of note, in TCGA-LIHC, all 25 genes were expressed at significantly higher levels in Imm-H than in Imm-L (FDR < 0.01; FC > 1.5) (Figure 2A).The immune scores were significantly higher in Imm-H than in Imm-L, consistently in the three datasets (P< 0.001) (Figure 2B).Furthermore,PD-L1, an antitumor immunosuppressive signature, was more highly expressed in Imm-H than in Imm-L (P< 0.01) (Figure 2C).Nevertheless, the ratios of immunostimulatory to immunosuppressive signatures (CD8+/CD4+ regulatory T cells), the base-2 Log-transformed values of the geometric mean expression levels of all marker genes of CD8+ T cells divided by those of CD4+ regulatory T cells, were significantly higher in Imm-H than in Imm-L (P< 0.05) (Figure 2D).Taken together, these results support that Imm-H has stronger anti-tumor immunity compared to Imm-L.
The immune-specific subtypes of HBV+ HCC have different clinical and phenotypic features
We compared 5-year OS and DFS prognosis between the immune-specific subtypes of HBV+ HCC in TCGA-LIHC, which had survival-related data available.Notably, Imm-H displayed significantly higher OS and DFS rates than Imm-L (P< 0.05) (Figure 3A).It supports the positive association between antitumor immune responses and survival prognosis in HBV+ HCC.We further compared several tumor progression-associated phenotypic features, including tumor stemness and ITH.We found that both stemness and ITH scores were markedly higher in Imm-L than in Imm-H in two of the three datasets (TCGA-LIHC and GSE121248) (P< 0.05) (Figure 3B).As expected, tumor purity was consistently higher in Imm-L than in Imm-H in the three datasets (Figure 3C).Altogether, these results indicate more favorable clinical outcomes in Imm-H than in Imm-L.
Comparisons of genomic and epigenomic features between the immune-specific subtypes of HBV+HCC
Tumor aneuploidy, also known as copy number alteration (CNA), is a typical genomic feature in tumors[23].We found that Imm-L had higher frequencies of arm-level copy number amplification and deletion across chromosomes (Figure 4A).Moreover, Imm-L likely had higher amplitudes of copy number amplification and deletion across chromosomes as compared to Imm-H (Figure 4B).It suggested a higher level of genomic instability in Imm-LvsImm-H, supporting a negative correlation between tumor aneuploidy and antitumor immunity[24].Nevertheless, TMB showed no significant difference between Imm-H and Imm-L (Mann-WhitneyUtest,P= 0.86).Furthermore, we found that Imm-H had significantly higher global methylation levels[25] than Imm-L (P= 0.006) (Figure 4C).It conforms to a previous study showing that reduced DNA methylation levels promote antitumor immunosuppression[25].
Pathways and GO enriched in the immune-specific subtypes of HBV+ HCC
Based on the DEGs between Imm-H and Imm-L, we identified the KEGG pathways enriched in Imm-H and Imm-L common across the three datasets.Because there were no pathways enriched in Imm-L overlapping among the three datasets, we only attained the pathways enriched in Imm-H.We identified a total of 39 pathways highly enriched in Imm-H common in the three datasets.As expected, many immune-related pathways were on the list, including allograft rejection, antigen processing and presentation, apoptosis, asthma, autoimmune thyroid disease, B cell receptor signaling, cell adhesion molecules, chemokine signaling, complement and coagulation cascades, cytokine-cytokine receptor interaction, cytosolic DNA sensing, Fc epsilon RI signaling, Fc gamma R-mediated phagocytosis, graftvshost disease, hematopoietic cell lineage, intestinal immune network for IgA production, Jak-STAT signaling, leishmania infection, leukocyte transendothelial migration, natural killer cell-mediated cytotoxicity, NOD-like receptor signaling, primary immunodeficiency, T cell receptor signaling, systemic lupus erythematosus, Toll-like receptor signaling, and viral myocarditis (Figure 5A).Besides, several stromal pathways were included in the list, such as ECM receptor interaction, focal adhesion, and regulation of actin cytoskeleton.Interestingly, we found two neuro-associated pathways included in the pathway list: neuroactive ligand-receptor interaction, and prion diseases.It indicates that the activities of these neuro-associated pathways are positively correlated with antitumor immunity.Indeed, the enrichment scores of these pathways correlated positively with immune scores in these datasets (Spearman correlation,P< 0.05) (Figure 5B).

Figure 1 ldentification of immune-specific subtypes of hepatitis B virus + hepatocellular carcinoma by clustering analysis.A: Based on the enrichment scores of 23 immune signatures, hierarchical clustering identifies two immune-specific subtypes of hepatitis B virus (HBV) + hepatocellular carcinoma (HCC): Imm-H and Imm-L, with high and low immunity, respectively, consistently in three datasets; B: Prediction of the immune-specific subtypes of HBV+ HCC by the Random Forest algorithm using the 23 immune signatures as attributes.The 10-fold cross-validation results in the training set and classification results in test sets are shown.
WGCNA[21] identified nine gene modules significantly differentiating HBV+ HCC by the immunespecific subtypes, OS, and/or DFS (Figure 5C).The gene modules upregulated in Imm-H while downregulated in Imm-L included the green module (with the representative GO term of immune response) and the yellow module (with the representative GO term of extracellular matrix) (P< 0.001).It is consistent with the results from the prior pathway analysis showing more highly enriched immune and stromal pathways in Imm-H versus Imm-L.Moreover, both modules were positively correlated with the OS prognosis (P< 0.05), consistent with the better OS in Imm-H relative to Imm-L.Besides, the blue module (with the GO term of homophilic cell adhesionviaplasma membrane adhesion molecules) was significantly and positively correlated with the OS prognosis (P= 0.02), although it showed no significant enrichment difference between Imm-H versus Imm-L.It is reasonable that the positive association between the blue module and OS since reduced homophilic cell adhesion can promote tumor progression[26].In contrast, the brown module had significant negative correlations with both OS and DFS time.The representative GO term for this module was cell cycle.It is justified since elevated cell cycle activity suggests tumor progression.
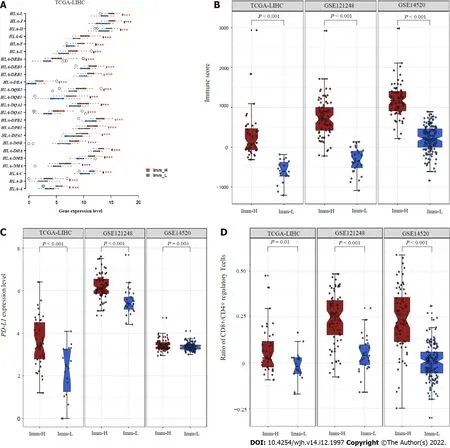
Figure 2 Comparisons of immune features between the immune-specific subtypes of hepatitis B virus + hepatocellular carcinoma.A: Imm-H shows significantly higher expression levels of HLA genes; B: Immune scores; C: PD-L1 expression levels; D: Ratios of immunostimulatory to immunosuppressive signatures (CD8+/CD4+ regulatory T cells) than Imm-L.The two-tailed Student’s t test P values are shown in (A, C, D), and the one-tailed Mann-Whitney U test P values are shown in (B).
Proteins enriched in the immune-specific subtypes of HBV+ HCC
We compared the expression levels of 219 proteins between the immune-specific subtypes of HBV+ HCC in TCGA-LIHC.We identified nine proteins differentially expressed between Imm-H and Imm-L (P< 0.05) (Figure 6A).Among them, MYH11, PDCD4, Dvl3, and Syk were more highly expressed in Imm-H, while PCNA, Acetyl-a-Tubulin-Lys40, ER-α_pS118, Cyclin E2, and β-Catenin were more highly expressed in Imm-L.As expected, the proteins more enriched in Imm-H showed significantly positive expression correlations with immune scores in HBV+ HCC, and the proteins more enriched in Imm-L showed negative expression correlations with them (Spearman correlation,P< 0.05) (Figure 6B).Previous studies have shown that most of these proteins have associations with tumor immune regulation.For example, MYH11 is a smooth muscle myosin of the myosin heavy chain family, whose expression has been associated with antitumor immune infiltration in cancer[27].PDCD4 is a tumor suppressor, whose expression in the tumor microenvironment is correlated with increased immune infiltration[28,29].Syk is also a tumor suppressor and has a role in tumor immune regulation[30].PCNA is involved in the DNA repair pathway in response to DNA damage, whose upregulation may promote tumor immune evasion[31,32].This is consistent with its upregulation in Imm-L versus Imm-H.ER-α has been shown to induce antitumor immunosuppression[33], supporting our finding.Cyclin E2 is a positive regulator of cell cycle, which may inhibit antitumor immune responses and immunotherapy responses[34].Again, this is consistent with our result.β-Catenin is an activator of the Wnt/β-catenin signaling pathway, whose overexpression suppresses antitumor immune responses[35], in line with our findings.
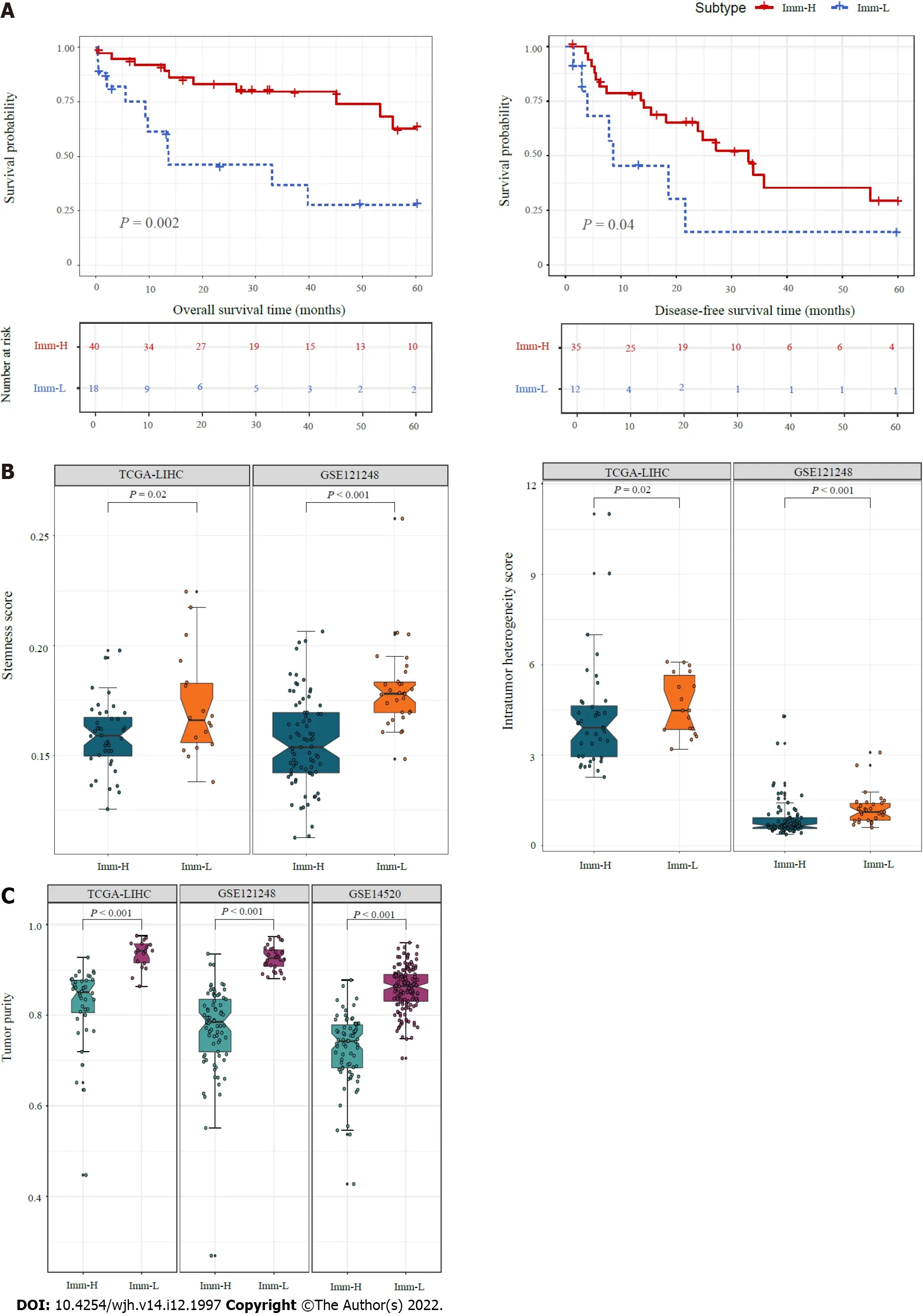
Figure 3 Comparisons of clinical and phenotypic features between the immune-specific subtypes of hepatitis B virus + hepatocellular carcinoma.A: K-M curves showing that Imm-H has significantly higher 5-year overall survival and disease-free survival rates than Imm-L.The log-rank test P values are shown; B: Imm-H has significantly lower stemness and intratumor heterogeneity than Imm-L; C: Imm-H has significantly lower tumor purity than Imm-L.The one-tailed Mann-Whitney U test P values are shown in (B, C).
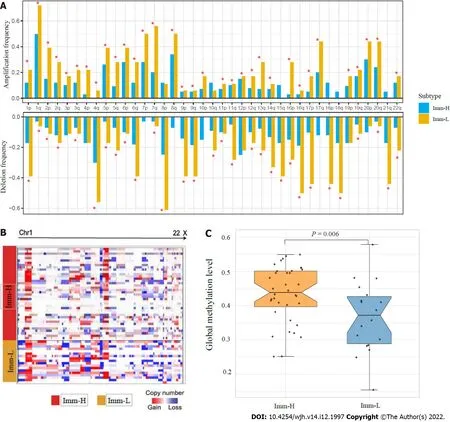
Figure 4 Comparisons of genomic and epigenomic features between the immune-specific subtypes of hepatitis B virus + hepatocellular carcinoma in TCGA-LlHC.A: Comparison of the arm-level SCNAs between Imm-H and Imm-L.The red asterisks indicate the chromosome arms in which Imm-L has higher amplification or deletion frequency than Imm-H; B: Heatmap showing that Imm-L likely has higher amplitudes of copy number amplification and deletion across chromosomes than Imm-H; C: Imm-H has significantly higher global methylation levels than Imm-L.The one-tailed Mann-Whitney U test P values are shown.
DlSCUSSlON
This study identified two immune-specific subtypes (Imm-H and Imm-L) of HBV+ HCC based on the enrichment of 23 immune signatures by unsupervised clustering.We showed that this subtyping method was reproducible as well as predictable by analyzing three different datasets.Furthermore, we demonstrated that both subtypes had significantly different clinical and molecular features.Compared to Imm-L, Imm-H displayed stronger immunity, more stromal components, lower tumor purity, lower stemness and ITH, lower-level CNAs, higher global methylation level, and better overall and disease-free survival prognosis (Figure 7).Our data support that “hot” tumors have a better prognosis than “cold” tumors in HBV+ HCC for their stronger antitumor immune responses.Similar findings were also observed in other cancers[36-38].Intriguingly, although continual inflammatory responses in thelivercaused by HBV infection is a major etiology for HBV+ HCC[39], higher immune/inflammatory responses are associated with a better prognosis in HBV+ HCC patients, as demonstrated by this analysis.It indicates that the relationship between immune/inflammatory responses and clinical outcomes in cancer is complex.Indeed, in some cancer types, such as glioma[40] and prostate cancer[41], the relationship between immune/inflammatory responses and clinical outcomes is negative.Thus, the relationship between immune responses and clinical outcomes in cancers is dependent on their tissue or cellular origins, the tumor microenvironment, the ratio of immunostimulatory over immunosuppressive signatures, as well as whether the immune response is the tumor progressionpromoting inflammation or immune cell-mediated elimination of tumor cells.

Figure 6 Proteins enriched in the immune-specific subtypes of hepatitis B virus + hepatocellular carcinoma in TCGA-LlHC.A: Nine proteins having significantly different expression levels between Imm-H and Imm-L.The two-tailed Student’s t test P values are shown; B: Spearman correlations between the expression levels of the nine proteins and immune scores in hepatitis B virus + hepatocellular carcinoma.The Spearman correlation coefficients (ρ) and P values are shown.
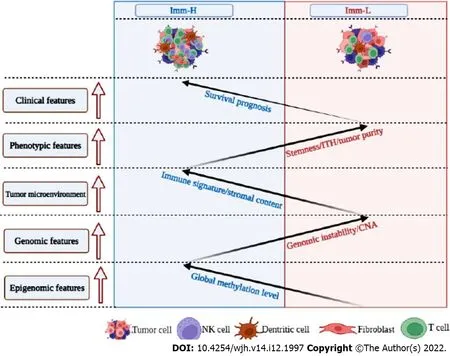
Figure 7 Schematic comparisons of clinical and molecular features between the immune-specific subtypes of hepatitis B virus + hepatocellular carcinoma.The figure was created with BioRender.com.
Prior studies have shown that TMB and CNAs have a positive and negative association with antitumor immune responses, respectively[24].However, our analysis suggests that TMB has no a significant association with antitumor immunity in HBV+ HCC, although CNAs have a negative association with antitumor immune responses.It suggests that it is CNAs but not TMB responsible for the significantly different immunity between the “hot” and “cold” tumor subtypes in HBV+ HCC.Furthermore, the significantly lower stemness and ITH of Imm-H compared to Imm-L suggest that stemness and ITH may lead to antitumor immunosuppression, consistent with previous findings[18,42,43].
Pathway analysis showed that two neuro-associated pathways (neuroactive ligand receptor interaction, and prion diseases) had higher enrichment in Imm-H than in Imm-L and that their upregulation was associated with increased tumor immune infiltration levels.The positive association between neuro-related pathways and antitumor immunity has been revealed in prior studies[44].Interestingly, many studies have demonstrated the inverse relationship between cancer and Alzheimer’s disease (AD)[45].AD is known as a progressive neurodegenerative disease as well as neuroinflammation disease[46].A recent study has proposed that AD is an autoimmune disorder of innate immunity[47].The present and prior data stimulate our imagination that the immune and inflammation could bridge the relationship between cancer and AD, such as hyperactivation of the immune system in AD patients reducing the risk of cancer.
Interestingly, besides the antitumor immune signatures, the immunosuppressive signature PD-L1, was also upregulated in Imm-HvsImm-L.Because both PD-L1 expression[48] and ample TILs[9] are positive predictors of the response to ICB, Imm-H would respond better to immunotherapy than Imm-L.Thus, our subtyping method may stratify HBV+ HCC patients for immunotherapy.That is, immunotherapy may yield morepropitiousefficacy for Imm-H than for Imm-L HBV+ HCC patients.
CONCLUSlON
HBV+ HCCs can be classified into two immune-specific subtypes in terms of their immune signature enrichment.Both subtypes have significantly different immunity, stromal contents, tumor purity, stemness, ITH, CNAs, methylation profiles, and survival prognosis.The immune-specific subtyping of HBV+ HCC may provide new biological insights as well as clinical implications for the management of this disease.
ARTlCLE HlGHLlGHTS
Research background
Hepatocellular carcinoma (HCC) is a major cancer of the liver that constitutes around 90% of liver cancer cases.Although traditional therapeutic approaches, including surgery, chemotherapy,radiotherapy, and targeted therapy, are effective in improving the survival of HCC patients, the overall survival prognosis of HCC patients is generally unfavorable.More recently, immunotherapy, such as immune checkpoint blockade, has achieved success in the treatment of various cancers, including HCC.However, only a small proportion of cancer patients respond well to immunotherapies to date.
Research motivation
Certain predictive markers for cancer immunotherapy responses have been uncovered, e.g., PD-L1 expression, tumor mutation burden (TMB), and mismatch repair deficiency.In addition, the tumor immune microenvironment (TIME) plays an important role in immunotherapy responses.Overall, the“hot” tumors infiltrated by a substantial number of tumor-infiltrating lymphocytes (TILs) are more responsive to immunotherapies, compared to the “cold” tumors lacking TILs.Hence, an investigation of the TIME in HCC would aid in the prediction of immunotherapy responses.
Research objectives
Despite these previous studies, the discovery of immune-specific subtypes of hepatitis B virus-positive(HBV+) HCC is worth investigating, considering that HBV infection is a major cause of HCC.
Research methods
In this study, to characterize the immunological landscape of HBV+ HCC, we identified its immunespecific subtypes by the unsupervised machine learning in transcriptomic data.Furthermore, we comprehensively compared the clinical and molecular features of these subtypes.
Research results
Compared to Imm-L, Imm-H displayed stronger immunity, more stromal components, lower tumor purity, lower stemness and intratumor heterogeneity, lower-level copy number alterations, higher global methylation level, and better overall and disease-free survival prognosis.
Research conclusions
Our immune-specific subtyping of HBV+ HCC may provide new biological insights as well as clinical implications for the management of this disease.
Research perspectives
This study is interesting for several reasons.First, for the first time, we identified immune-specific subtypes of HBV+ HCC based on immune signature scores and demonstrated that this new subtyping method was reproducible in three different datasets.Second, our subtyping method captures the comprehensive heterogeneity of HBV+ HCC in the tumor microenvironment, genomic integrity, protein expression profiles, DNA methylation profiles, tumor stemness, intratumor heterogeneity, and clinical outcomes.Third, our data suggest that it is copy number alterations but not tumor mutations responsible for the different immunity between the “hot” and “cold” tumor subtypes in HBV+ HCC.Finally, our identification of the immune-specific subtypes of HBV+ HCC may provide new insights into the tumor biology and identify the HBV+ HCC patients beneficial from immunotherapy.
FOOTNOTES
Author contributions:Li SW contributed to software, validation, formal analysis, investigation, data curation, visualization, writing - review & editing; Han LF contributed to software, formal analysis, data curation; He Y contributed to data curation; Wang XS contributed to conceptualization, methodology, resources, investigation, writing - original draft, supervision, project administration, funding acquisition.
lnstitutional review board statement:Because we did not perform any human/animal experiments in this research, we could not provide the following file: Institutional review board approval form or document.
lnstitutional animal care and use committee statement:Because we did not perform any animal experiments in this research, we could not provide the file.
Conflict-of-interest statement:All the authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data sharing statement:No additional data are available.
ARRlVE guidelines statement:The authors have read the ARRIVE guidelines, and the manuscript was prepared and revised according to the ARRIVE guidelines.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial.See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORClD number:Yin He 0000-0003-2460-6472; Xiao-Sheng Wang 0000-0002-7199-7093.
S-Editor:Liu JH
L-Editor:A
P-Editor:Liu JH
杂志排行

World Journal of Hepatology的其它文章
- Role of microRNA-regulated cancer stem cells in recurrent hepatocellular carcinoma
- Liver chemistries in severe or non-severe cases of COVlD-19: A systematic review and meta-analysis
- CLlF-SOFA and CLlF-C scores for the prognostication of acute-onchronic liver failure and acute decompensation of cirrhosis: A systematic review