Comparative population genomics reveals glacial cycles to drive diversifications in tropical montane birds(Aves,Timaliidae)
2023-01-03PerEricsonMartinIrestedt
Per G.P.Ericson,Martin Irestedt
Department of Bioinformatics and Genetics,Swedish Museum of Natural History,P.O.Box 50007,SE-10405,Stockholm,Sweden
Keywords:Aves Timaliidae Babblers Demographic history Fragmented distributions Pleistocene glacial cycles Population genomics Tropical mountain forests
ABSTRACT Many bird species are specialized to live in the broadleaved,evergreen forests in the mountain regions in Southeast Asia.These mountain habitats are not continuously distributed as the different mountain areas are separated by lowlands,which has restricted gene flow and thus contributed to the high biological diversity in this region.The degree of connectivity between mountain areas has fluctuated with the Pleistocene glacial cycles,being largest during the glaciations when the mountain forests spread to lower elevations.Here we study how the intermittent periods of restricted gene flow and connectivity between the populations of five montane species of babblers(Aves,Timaliidae)in Vietnam may be traced in their genomes.The results suggest that the babbler species in the Central Highlands have been isolated from their sister-populations in northern Vietnam for between ca.585 and 380 ky.For two species with populations in both the Central Highlands and the Da Lat region,we found that these split at more or less the same time(440–340 kya).We also found a significant statistical correlation between the time of the splits of these populations and the lowest altitude at which they are known to occur(no similar correlation was found with the geographic distances between populations).The populations in northern Vietnam show higher genetic variation than their counterparts in South-Central Vietnam,supporting the postulate that smaller populations may have lower genetic variation than larger.In accordance with this,we found the lowest genetic variation in the two species with the smallest populations in the Central Highlands.These two populations also show low levels of genomic heterozygosity.Our results show that the south-central populations of the studied babbler species are genetically distinct from their sister-populations in northern Vietnam,providing additional argument for the long-term protection of the evergreen mountain forests in Southeast Asia.
1.Introduction
Pleistocene climate fluctuations have shaped the patterns of genetic diversity observed in many extant species(Taberlet et al.,1998;Avise,2000;Hewitt,2004;Himes et al.,2008).This is especially prominent in mountain habitats where the glacial cycles have expanded and contracted suitable environments along an altitudinal gradient leading to alternating periods of genetic isolation and connectivity of populations(Hewitt,1996,2000,2004;Wiens,2004;Qu et al.,2011;Steinbauer et al.,2016;Camacho-Sanchez et al.,2018).This process has largely promoted and preserved genetic diversification of mountainous species,which is considered an important explanation of why mountain regions are exceptionally rich centers of biodiversity and endemism(Kier et al.,2009).Although the glacial cycles have influenced gene flow and effective population size in most montane species,their responses may vary because of species-specific differences in inter-acting factors such as ecology,demography and local topography(Avise,2000).Most research on the interaction between glacial cycles and genetic diversity has focused on northern latitudes,while species living in tropical and subtropical mountain habitats are still under-studied.
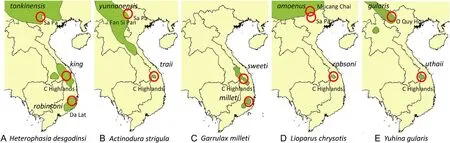
Fig.1.Geographic distributions of five species of babblers with allopatric populations in Southeast Asia.The red circles show the localities for the samples included in this study.The distributions are based on range polygons obtained from BirdLife International and Handbook of the Birds of the World(2020).(For interpretation of the references to colour in this figure legend,the reader is referred to the Web version of this article.)
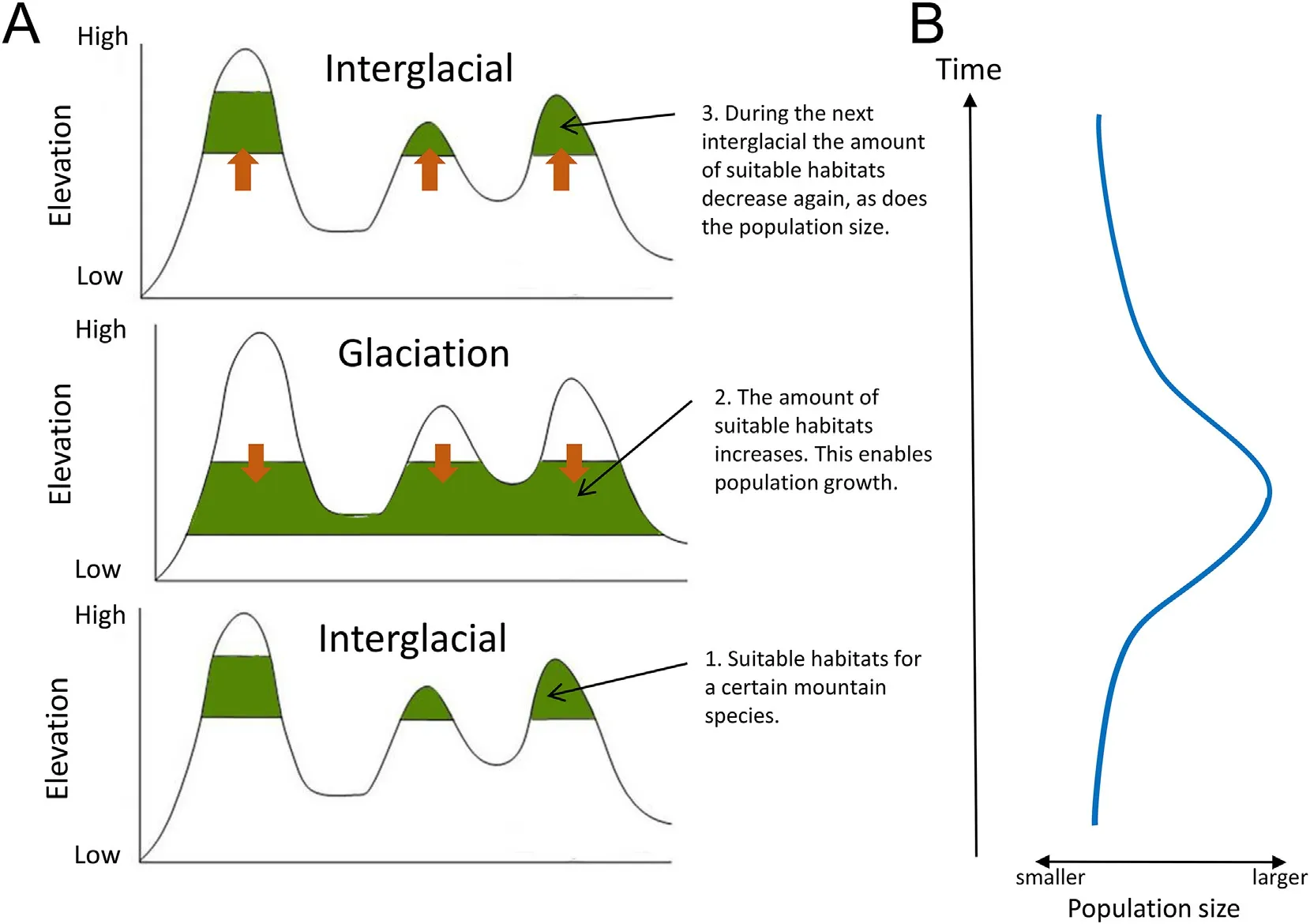
Fig.2.Schematic illustration of how the glacial cycles may have affected a mountain habitat and the species adapted to it.During a cold glacial,the mountain habitats are pushed downhill(A),by which their distribution area increases in size(B).The increased amount of suitable habitats allows the population sizes of species to increase.During the warmer interglacials,both the amount of suitable mountain habitats and the size of the populations decrease.
The mountains in Southeast Asia house an extraordinary high biodiversity,unique endemism,and include one of the largest continuous natural forest areas in continental Asia(https://www.wwf.org.la/projec ts/carbi/).The mountains in northern Vietnam represent the far eastern part of the Himalayan uplift,from where the Annamite Mountain Range stretches south to the Central Highlands and the Da Lat Plateau(Nam,1995;Dinh et al.,2022).The mountains are mostly covered with broadleaf evergreen forests and consist of a number of separate massifs,some with peaks>2500 m tall.Between these massifs are regions with lower elevations where the habitats are unsuitable for animals and plants adapted to higher elevations.Consequently,many montane species have disjunct distributions in Southeast Asia.Some typical examples are babblers(Aves,Timaliidae),i.e.,Heterophasia desgodinsi(Black-headed Sibia),Actinodura strigula(Chestnut-tailed Minla),Garrulax milleti(Black-hooded Laughingthrush),Lioparus chrysotis(Gold-breasted Fulvetta),and Yuhina gularis(Stripe-throated Yuhina),which occur at elevations above 2000 m in three major mountain regions that are separated by lowland habitats(Fig.1).These species have rather restricted,high-elevation distributions in the Central Highlands and the Da Lat Plateau,and they also have sister-populations living in northern Vietnam and southern China(Fig.1).Furthermore,all populations in South-Central Vietnam have been separated from their northern sister-populations long enough to develop distinct plumage characters that have been used to describe them as subspecies(Eames,2002).The case of Garrulax milleti is similar although this is an endemic species restricted to South-Central Vietnam.
The five babbler species studied herein all have rather small populations in the Central Highlands.They were all discovered after fieldwork in the 1990's,during which a limited number of voucher individuals were collected(Eames,2002).Although relatively rare,these species contribute to uniqueness of tropical mountainous endemism.In particular,as habitat loss is a major threat to many organisms throughout Southeast Asia,we believe more information about the phylogeography of these little-known birds in the Central Highlands will be useful in the protection of the evergreen mountain forests upon which they depend.
We postulate that the glacial cycles during the Pleistocene have affected the distributions of these high elevation species.During the glaciations,evergreen mountain forests are pushed to lower altitudesleading to an increase of the area of suitable habitats available for the species adapted to them(cf.He and Jiang,2014;Colwell et al.,2008),such as the babbler species studied here(Fig.2).This meant the population sizes of these species could increase and although their distributions may never have been continuous,the geographic distances between the different populations probably decreased considerably.Thus,we would expect to find that the sizes of the populations have fluctuated in parallel with the glacial cycles,and we hypothesize that this is observable in the genomes of these individuals.We also postulate that the populations became fragmented during warm interglacial period when the amount of suitable habitats was the smallest.
2.Material and methods
2.1.Study system and sampling
For our study,we have used samples of five species of babblers(Aves,Timaliidae)living in the highlands of Southeast Asia,Garrulax milleti,Lioparus chrysotis,Heterophasia desgodinsi,Yuhina gularis and Actinodura strigula.All samples were obtained from the BirdLife International Vietnam Programme and the Swedish Museum of Natural History.Only few specimens of birds have been collected in the Central Highlands,but we were allowed to use toepads of individuals collected in the late 1990's by Jonathan Eames,Nguyen Cu and colleagues(Eames,2002).There are also few individuals available in museum collections of the populations in the Da Lat Plateau(Eames and Ericson,1996),and we used toepads from study skins to sequence these populations too.For the populations in northern Vietnam,however,we were able to include fresh samples collected during fieldwork conducted between 1994 and 2012 by the Swedish Museum of Natural History and the Institute of Ecology and Biological Resources,Hanoi.
2.2.Generation of data,processing,and variant calling
DNA from the frozen samples was extracted using the KingFisher duo extraction robot and the KingFisher™Cell and Tissue DNA Kit according to the manufacturer's instructions.Genome library preparation(using Illumina TruSeq DNA Library Preparation Kit)for these samples was performed by SciLifeLab.For laboratory procedures and genome library preparation from toepad samples see Meyer and Kircher(2010)and Irestedt et al.(2022).All libraries were sequenced on Illumina Nova-Seq6000 or Illumina HiSeq X platforms.The raw reads have been deposited at the NCBI Sequence Read Archive(SRA)(Accession No.PRJNA880318).
Illumina sequencing reads were processed using a custom-designed workflow(available at https://github.com/mozesblom/nf-polish)to remove adapter contamination,low-quality bases and low-complexity reads.Overlapping read pairs were merged using PEAR(Zhang et al.,2014)and SuperDeduper(Petersen et al.,2015)was used to remove PCR duplicates.Trimming and adapter removal were done with TRIMMOMATIC v.0.32 with default setting(Bolger et al.,2014),and low-complexity reads were removed using a custom-made python script.Overall quality and length distribution of sequence reads were inspected with FASTQC v.0.11.5(Andrews,2010)before and after the cleaning.DNA degradation patterns in historical specimens could lead to a consistent signal of erroneous substitutions.As degradation almost exclusively occurs at the ends of DNA-fragments we first investigated how the trimming of reads would affect the number of recovered variants.We compared the results after trimming 5 bp and 10 bp from both ends,respectively.We found that the number of variants called drops considerably after the trimming of the ends,but we found almost identical results after removing 5 bp as after 10 bp.We thus based all analyses herein on reads from which 5 bp were removed from both ends.
We used BWA mem v0.7.12(Li and Durbin,2009)to map the cleanedreads against the Mixornis gularis(Pin-striped Tit-Babbler)genome(Tan et al.,2018;GenBank GCA_003546035).The mapping coverage varied from 5×to 30×(Table 1).High-quality SNPs(single-nucleotide polymorphisms)were called from the BAM-files using mpileup in Samtools v1.4 and bcftools v1.5(Li et al.,2009;Danecek et al.,2021)to include individual biallelic genotypes with a minimum depth of 10 reads per individual and a minimum variant quality of 30.We also filtered variants with a minor allele frequency(MAF)of 10%or less(i.e.,at least two out of ten reads support either of the two alleles).We called SNPs for each species separately and filtered the datasets to include only SNPs that could be scored for all individuals(Appendix Table S1).
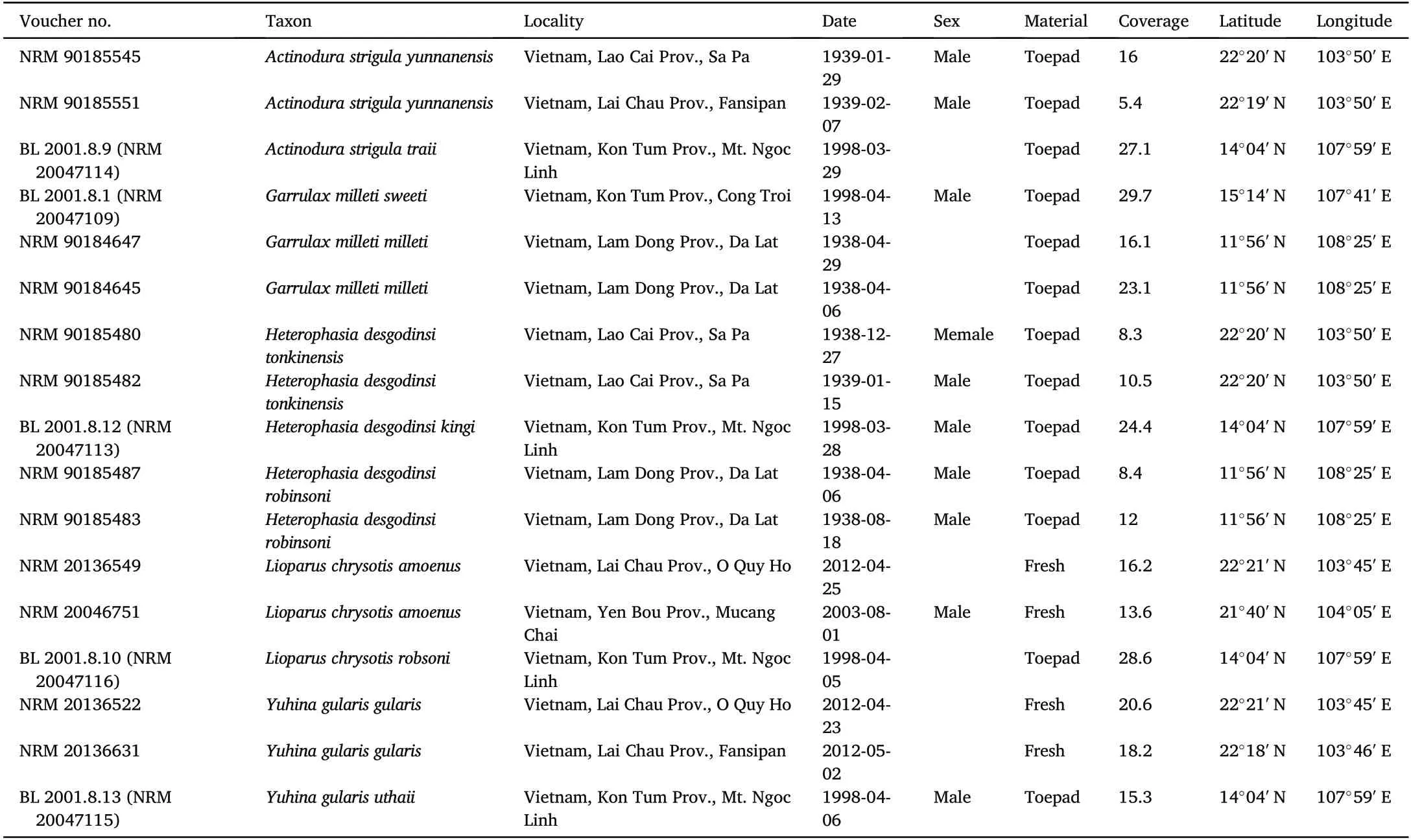
Table 1 Samples used in the study.
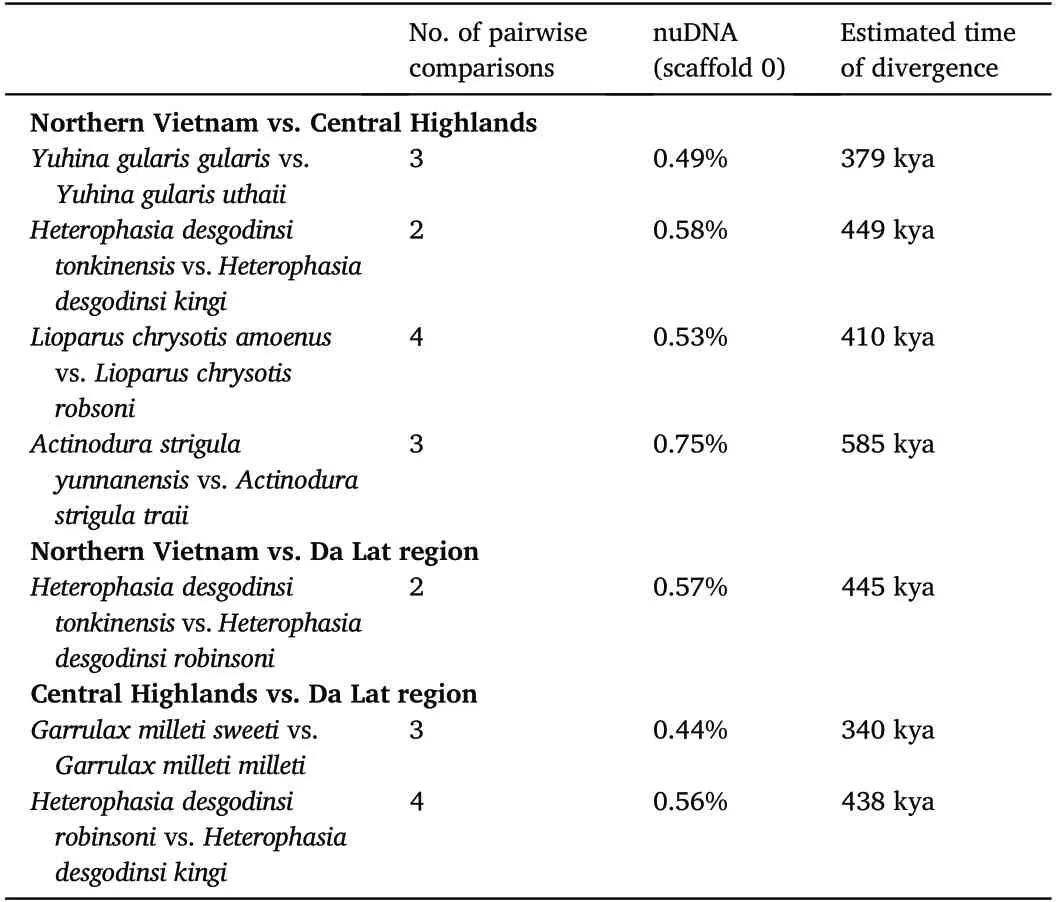
Table 2 Mean values for the observed genetic distances and the estimated divergence time between the populations in northern Vietnam and those in the Central Highlands and Da Lat region in the different babbler species.
2.3.Demographic history and divergence time
To evaluate the influence on geography on the genetic structure across the five babbler species we estimated the genome-wide,withinspecies divergence between the individuals as the pairwise p-distances based on an alignment of scaffold 0(19.5 Mbp).The observed genetic distances were correlated across species with the lowest elevations at which these species are known to occur and the shortest geographic distance between their respective intra-specific populations.Data on elevations were obtained from Collar and Robson(2020),while geographic distributions were downloaded as range polygons from BirdLife International and Handbook of the Birds of the World(2020).The digitalized distributions were imported in GoogleEarth,which was used to measure the distances between the allopatric populations.
We estimated the timing of the splits between populations by translating the observed genetic distances into years by using a mean average rate of divergence between two taxa of 0.128% per million years(Ellegren,2007).
We used the pairwise sequentially Markovian coalescent(PSMC)model(Li and Durbin,2011)to estimate changes in effective size of the different populations through time.PSMC utilizes the fact that each diploid genome is a collection of hundreds of thousands independent loci.By estimating the time to the most recent common ancestor(TMRCA)of the two alleles at each locus a distribution of TMRCA across the genome is created.We set the recombination rate to 1.0×10-8,the genomic mutation rate per generation to 4.6×10-9(Smeds et al.,2016),and we used the estimates by Bird et al.(2020)of the generation length for each species.We followed the recommendations by Nadachowska-Brzyska et al.(2015)and set the parameters for the PSMC analysis to-N30-t5-r5-p“4+30×2+4+6+19”.We also overlaid the curves for the populations in the Central Highlands and adjusted them along the x-axis(by adjusting the generation lengths used in the calculations)to make them fit the hypothesis that the population fluctuations are related to the availability of suitable habitats.We then compared these manually adjusted generation lengths with those published by Bird et al.(2020).
Data from the Vostok and EPICA ice cores(downloaded from www.climatedata.info)was used as proxy for changes in the global temperature during the last 800 ky.
3.Results
The observed genetic distances between the populations in northern Vietnam and those in the Central Highlands and the Da Lat Plateau,vary between 0.44% and 0.75%(Table 2).Translated to calendar years this suggests that the population of Actinodura strigula may have been isolated in the Central Highlands for as long as 585 ky,while the latest separation between populations occurred in Yuhina gularis ca.379 kya(Table 2).For the two species with populations in both the Central Highlands and the Da Lat region,these split occurred at ca.438 kya(Heterophasia desgodinsi)and 340 kya(Garrulax milleti).

Fig.4.(A)Estimates of genome-wide heterozygosity in the different babbler species show that the populations in South-Central Vietnam have an on average lower degree of heterozygosity than the populations in northern Vietnam.(B)The PSMC curves indicate that the populations of Yuhina gularis and Actinodura strigula in South-Central Vietnam always have been much smaller than the corresponding populations further north.This pattern is not observed in the other species.
We found a positive correlation(r=0.72,p<0.01)between intraspecific genetic distance and the lowest elevations where these species occur across all these five babbler species(Fig.3).We also found a significant correlation(r=0.62,p<0.01)between the genetic distance and the mid-elevation for the distribution of the respective species(Appendix Fig.S1).However,we did not find significant correlation between intraspecific genetic distance and the shortest geographic distance between these allopatric populations(Appendix Fig.S2).These results suggest that the intraspecific divergence of the five species is likely due to elevation-dependent historical processes rather than to allopatric isolation alone.
We found,however,that the genetic variation is on average lower in individuals representing the South-Central Vietnam populations than in those sampled from their northern sister-populations(Fig.4;Appendix Table S2).Furthermore,the lowest genetic variation was found in the individual of Actinodura strigula in the Central Highlands,which represents a population with one of the smallest geographic distributions studied here(Fig.1).
We compared the degree of heterozygosity in the individuals of Lioparus chrysotis,Heterophasia desgodinsi,Yuhina gularis and Actinodura strigula collected in northern and South-Central Vietnam and found a lower degree of heterozygosity in these than in their conspecific representatives of the South-Central Vietnam populations.The largest differences were found in Yuhina gularis and Actinodura strigula.In both Garrulax millet and Heterophasia desgodinsi the individuals in the Central Highlands and in the Da Lat Plateau show similar degrees of heterozygosity.
The PSMC curves of the temporal fluctuations in effective population size are roughly bell-shaped for all individuals.The general pattern is that they indicate a steadily increase in the respective population through the Pleistocene until it reaches a maximum size after which it decreases.Another increase in population size was observed at the very end of Pleistocene in a few individuals.When comparing the curves individually it is obvious that the ones for Yuhina gularis and Actinodura strigula resemble each other.The curves for the other species give a more complex picture of the fluctuations in effective population size over time,although most species show a decline in population size after ca.100 kya.When inferring the maximum Pleistocene population sizes of the populations in the Central Highlands and the Da Lat Plateau from the individual PSMC curves,we found Yuhina gularis and Actinodura strigula to have the by far smallest populations of the five babbler species.Furthermore,the sizes of the Heterophasia desgodinsi and Garrulax milleti populations in the Da Lat Plateau were estimated to be considerably smaller than those in the Central Highlands.
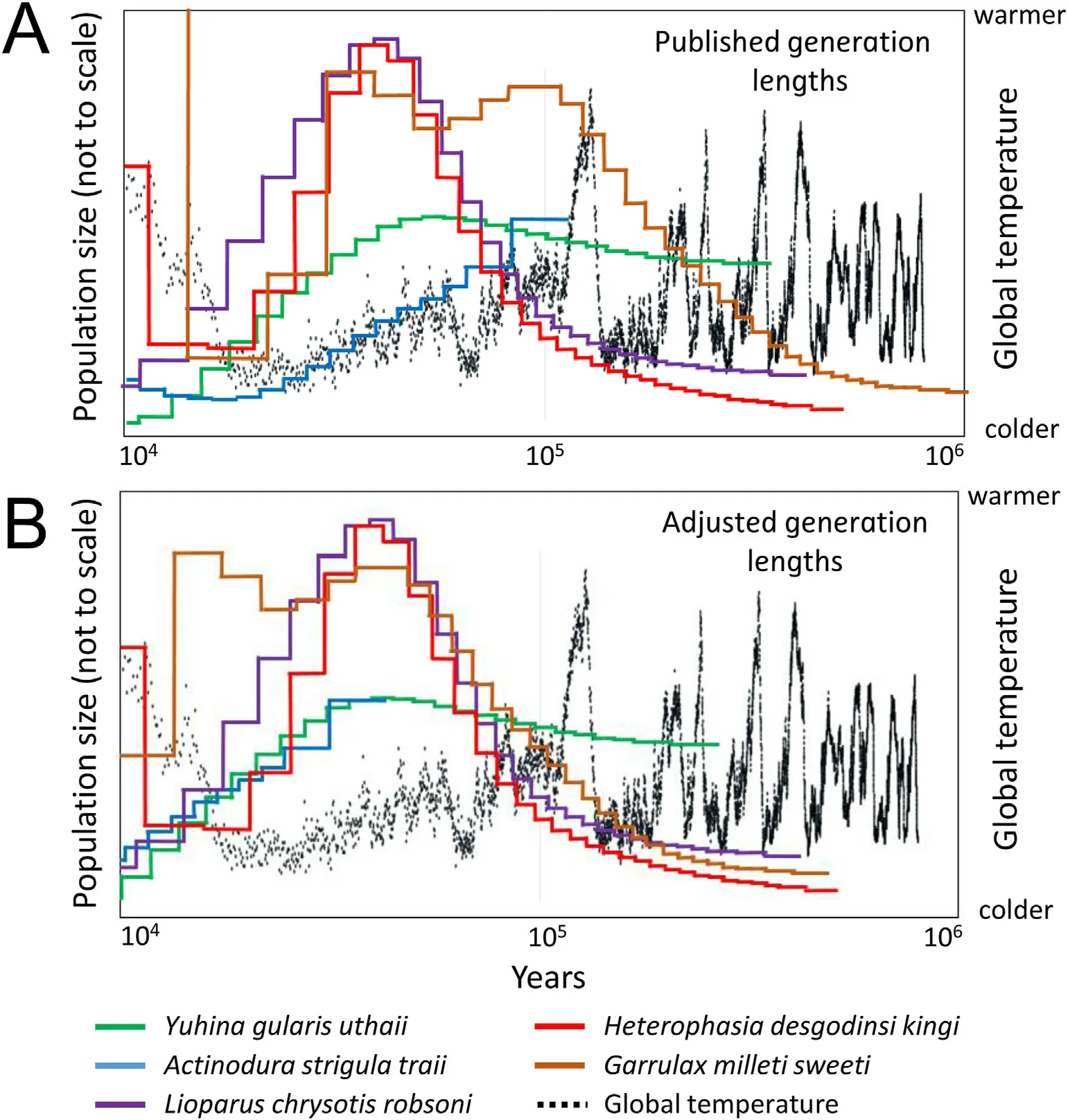
Fig.5.(A)PSMC curves for the populations in the Central Highlands based on the generation lengths published by Bird et al.(2020)superimposed on the estimated fluctuations in global temperature during the glacial cycles.The size fluctuations of the five populations seems not to co-vary with temperature as would be expected if they were affected by the glacial cycles in a similar way.However,we may assume that the population sizes were largest during the glaciations(when the larger areas of suitable habitats for these species were available)and that the fluctuations in the curves should show this.We can then shift the position of the PSMC curves sideways by changing the generation length used in the calculations.In(B)the generation lengths have been adjusted so that the maxima of the curves coincide at approximately the same time,ca.50 kya.The generation lengths required to obtain this figure are given in Appendix Table S3.
4.Discussion
The mountain fauna of South-Central Vietnam shows a high degree of endemism.Besides several new taxa of birds(Eames et al.,1999a,1999b;Eames,2002),some of which are included in this study,many other vertebrate species new to science have been discovered in the Central Highlands(e.g.,Orlov,2005;Abramov et al.,2006;Kruskop et al.,2006;Jenkins et al.,2007).The large number of endemic vertebrates found suggests that these mountain habitats have been isolated from other mountain regions during a long time.The results herein suggest that the babbler species in the Central Highlands have been isolated from their northern sister-populations for between ca.585 and 380 kya.The estimates for the two species with populations in both the Central Highlands and the Da Lat region show that these split at more or less the same time(440–340 kya).Since their separation,the populations in South-Central Vietnam have evolved enough phenotypic differences to merit them taxonomic rank as subspecies(Eames,2002).
It is likely that the temporal fluctuations in effective population size observed in the five species of babblers living in the mountain regions in Southeast Asia have been caused by shifting climatic conditions during the Pleistocene glacial cycles.During the glaciations,the broadleaved evergreen mountain forests were pushed to lower elevations(cf.He and Jiang,2014;Colwell et al.,2008),which led to an increase of their distribution area.This,in turn,would allow populations adapted to these forests to increase in size.Likewise,when the warmer climate conditions during the interglacials pushed montane forests to higher elevations,the babbler populations decreased in size and their distributions were fragmented.All populations are today widely separated(the geographic distances in the different species range from 320 to 1040 km)with no suitable habitats between the respective mountain region.A recent exchange of individuals thus seems unlikely.
Given that the suitable habitats for these montane species have been pushed up and down in altitude in response to the changing climate during the glacial cycles,we postulated that the lowest altitude occupied by the populations would correlate positively with the time of separation between the populations in northern and South-Central Vietnam.We used the observed genetic distance as proxy for the time of separation and we did find a significant statistical correlation with the lowest altitude.However,we found no similar correlation with the geographic distances between the populations.The results suggest that the elevational fluctuations in distribution during the glacial cycles have influenced the genetic structuring between the populations in northern and South-Central Vietnam of these babbler species.Although we find a shared,inter-specific genetic pattern linked to the elevational distributions of these species,isolation-by-distance is only one of the factors that explains the observed differences in the genetic structure between them.The factors determining much of the remaining genetic structure among the species is surely due to differences in species-specific conditions.
The observation that the populations in northern Vietnam all show higher genetic variation than their counterparts in South-Central Vietnam supports the postulate that smaller populations may have lower genetic variation than larger.In accordance with this,we found the lowest genetic variation in Yuhina gularis and Actinodura strigula that have the smallest populations among the studied babbler species in the Central Highlands(as inferred from the PSMC curves).These two populations also show low levels of genomic heterozygosity.
When superimposing the PSMC curves for the populations in the Central Highlands,we found that the timing for their maximum size varies among the species(Fig.5A).Under the assumption that the populations reached their maximum size when the amount of suitable habitats were the largest,we would expect these curves to reach their maxima at approximately the same time.When plotting the results of the PSMC analysis the curve can be shifted along the x-axis(i.e.,the timescale)by changing the generation length used to scale the plot.In Fig.5A we used the generation lengths estimated by Bird et al.(2020).Note that the generation length refers to the average age of the breeding individuals in the population.In order to get a robust estimate of generation length one needs information on life-history traits such as maximum longevity,age at first reproduction,and annual adult survival,parameters that are virtually unknown for most wild bird populations.Most estimates published by Bird et al.(2020)are based on interpolations or imputations of data observed for other species.Assuming that the population sizes did reach their maxima at about the same time,we may estimate what generation times are required to align the curves in relation to the temperature curves since the last interglacial(as shown in Fig.5B).It turns out we need to use slightly shorter generation lengths for Yuhina gularis,Actinodura strigula and Garrulax millet than those estimated by Bird et al.(2020)(Appendix Table S3).We want to stress that this naive way to estimate generation lengths“backwards”by fitting PSMC curves to known temperature fluctuations,is applied here merely to find out if such an approach would require using unrealistic generation lengths.Although the three new estimates of generation lengths all are shorter than those published,we do not regard them as unreasonably short.Ideally,a better future knowledge of the life-history of these species will result in more reliable estimates of their demographic history.
The habitat loss is a major problem to the survival of many populations with small populations.This is not least true for the evergreen mountain forests in Southeast Asia.Our results show that the five studied babbler species are genetically distinct from their sister-populations in northern Vietnam and thus are of large value to conservation.Although the results may be regarded as tentative because of the limited sampling of individuals,we hope they will contribute to the appreciation of the high biological value of these mountain habitats and provide additional arguments for their long-term protection.
Authors’contribution
PE conceived and designed the study.MI performed the genome sequencing and PE the comparative population genomic analyses.PE drafted the manuscript with assistance from MI.Both authors read and approved the final manuscript.
Ethics statement
All research in this paper is based on pre-existing museum collections that have been collected under appropriate permits over many decades.
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:Per G.P.Ericson reports financial support was provided by Swedish Research Council.
Acknowledgements
Samples for this study were kindly provided by the Swedish Museum of Natural History and the BirdLife International Vietnam Programme.We are particularly grateful to Jonathan Eames,Ulf Johansson and Peter Nilsson for assisting with sampling of study skins in their care,and to Yanhua Qu for commenting on an earlier draft of this manuscript.We also want to thank Nguyen Cu and the Institute of Ecology and Biological Resources in Hanoi for logistic support during fieldwork in Vietnam.We acknowledge support from Science for Life Laboratory,the National Genomics Infrastructure(NGI)and Uppmax for providing assistance in massive parallel sequencing and computational infrastructure.This work was supported by the Swedish Research Council(grant no.621-2017-3693 to PE).
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://do i.org/10.1016/j.avrs.2022.100063.
杂志排行

Avian Research的其它文章
- Identification of annual routines and critical stopover sites of a breeding shorebird in the Yellow Sea,China
- Differing migration patterns and year-round habitat-use of allopatric nesting Eastern Tundra Bean Geese(Anser fabalis serrirostris)in East Asia
- GPS tracking data reveal the annual spatiotemporal movement patterns of Bridled Terns
- Ecological traits affect the seasonal migration patterns of breeding birds along a subtropical altitudinal gradient
- Demographic and behavioral responses of the White-winged Dove to human disturbances during winter in northwestern Mexico
- Urban avifauna distribution explained by road noise in an Eastern European city