电感耦合等离子体发射光谱(ICP-OES)法测定藏药原材料中的砷汞
2023-01-01谢海东鲁海妍
谢海东 鲁海妍 张 炜
(1 青海省地质矿产测试应用中心,西宁 810021;2 青海省药品检验检测院,西宁 810000)
藏药是在广泛吸收融合了中医药学、印度医药学和大食医药学等理论的基础上,通过长期实践所形成的独特医药体系,迄今已有上千年的历史,是我国较为完整、较有影响的民族药之一。青海占据着青藏高原北部和东部地区的广大地区,省内有药用资源1 294种,其中植物类1 087种、动物类150种、矿物类57种。全省常用的几百种藏药中有70%采自青藏高原。
据史料记载,公元前200多年,一位叫孜拉嘎玛跃德的人提出“有毒就有药”的观点。说明了毒可成药、以毒攻毒的医理。在藏药各名贵成分中,“佐苔”是历代名藏医通过对剧毒水银特殊炮制加工成的无毒、具有奇特疗效的药品,被雪域人民称之为藏药中的至宝。为了统一藏药的配制,西藏及青海、甘肃、四川、云南、新疆等六省区于1977年分别在拉萨和西宁召开会议,对174种单科药和290种配方进行定量,为以后藏药的生产、使用提供科学依据。因此准确测定藏药原材料中的砷、汞是很有必要的。
一般来说,砷、汞的检测常用原子荧光光谱法[1-2]、原子吸收光谱法[3-4]、电感耦合等离子体质谱法[5-6]等,这些方法较适合砷、汞含量较低的土壤、水系沉积物、食品、农产品、化妆品等的测定。而藏药原材料中砷、汞含量较高,有的主要成分是雄黄和雌黄,有的甚至肉眼可见水银珠,上述方法已无法满足测试要求,而传统的容量法[7]流程长,速度慢,使用试剂多,且两种元素需单独滴定分析,不适合批量分析。20世纪70年代后,电感耦合等离子体发射光谱(ICP-OES)技术取得了很大进展,因其具有检出限低、精密度高、准确度好、线性范围宽、分析速度快、基体效应小、同时可测定多种元素等优点,被普遍应用于多元素的分析[8-9]。近年来,采用电感耦合等离子体原子发射光谱法测定砷等元素也有不少报道[10-13],但采用电感耦合等离子体发射光谱法测定汞还鲜见报道。本文采用微波消解对样品进行前处理,选择ICP-OES测定藏药原材料中的砷、汞,通过检出限、精密度、加标回收、方法比对等试验,证实该方法测定藏药原材料中砷、汞的可行性。
1 实验部分
1.1 仪器设备及工作条件
iCAP6300电感耦合等离子体发射光谱仪(美国赛默飞世尔科技公司),AL104分析天平(梅特勒-托利多仪器有限公司),ETHOS UP型微波消解仪(意大利迈尔斯通公司),VB48UP型赶酸仪(莱伯泰科仪器股份有限公司),101-3A电热干燥鼓风箱(常州澳华仪器有限公司);高纯氩气,纯度99.9%。仪器测量条件见表1。

表1 iCAP仪器测量条件
1.2 主要试剂材料
盐酸(ρ=1.19 g/mL,白银良友化工有限公司,GR),硝酸(ρ=1.42 g/mL,白银良友化工有限公司,GR),过氧化氢(AR,天津百世化工有限公司)。
混合消解液:取40 mL硝酸与10 mL过氧化氢混合,摇匀。
重铬酸钾(10 g/L):称取1 g重铬酸钾溶于100 mL水中,摇匀。
砷标准储备溶液(1.0 mg/mL):准确称取1.320 2 g三氧化二砷(基准预先在100~105 ℃烘1 h,置于干燥器中冷却至室温)于100 mL烧杯中,加入20 mL氢氧化钠溶液(ρ=200 g/L),低温加热使其溶解,加30 mL水,加2滴酚酞指示剂(1 g/L),用硫酸中和至红色刚好消失,再过量2 mL,冷却后,移入1 000 mL容量瓶中,用水稀释至刻度,摇匀。
汞标准储备溶液(1.0 mg/mL):准确称取1.354 0 g预先用P2O5干燥24 h的二氯化汞,少量水溶解,加入50 mL硝酸(ρ=1.42 g/mL)、10 mL重铬酸钾溶液(10 g/L),用水定容至1 000 mL,混匀。
实验用水为离子交换方法制备的二级水,电阻率≥18.2 MΩ·cm。
1.3 标准曲线的绘制
以砷标准储备溶液和汞标准储备溶液为母液,逐级稀释相对应的As、Hg混合标准系列浓度分别为0.50、1.0、2.0、20、50、100、200 μg/mL,以王水(10+90,V/V)为介质。
1.4 样品预处理
准确称取试料0.10 g(精确至0.000 1 g)于微波消解管中,加入混合消解液8 mL,摇散样品,旋紧管塞,在室温下过夜预消解,次日于微波消解仪内按表2步骤进行消解,冷却35 min后取出。
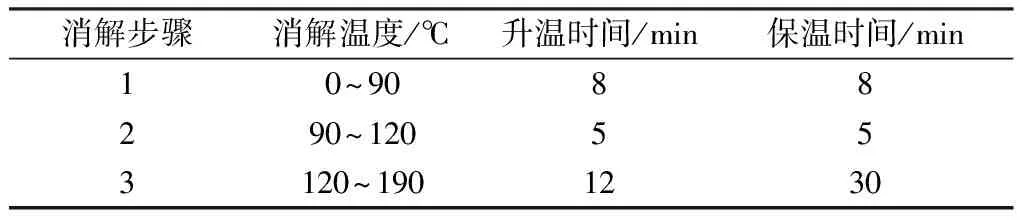
表2 消解步骤
消解完毕,待消解仪自动降温至室温时,取出消解管,缓慢泄压放气,小心将消解管置于赶酸仪内蒸至小体积(约1~2 mL)时取下,加入10 mL王水(1+1,V/V)提取,转移至100 mL容量瓶中,多次洗涤消解管,合并洗涤液,补加10 mL王水(1+1,V/V),用水定容至刻度,摇匀,澄清待测。
2 结果与讨论
2.1 样品预消解
实验所选藏药原材料不但含有较高砷、汞,而且含有大量有机物,如果直接进行微波消解,有机物被氧化后可形成二氧化碳等大量气体,在反应管中形成较大压力,如果使用自泄压反应管,将导致频繁泄压,本实验采用全密闭反应管,若管内压力过大,可能导致防爆膜破裂。因此,实验选择预消解24 h,将容易消解的部分先反应掉,减少在微波消解过程中产生的压力。
2.2 消解试剂的选择
准确称取0.10 g(精确至0.000 1 g)国家Ⅰ级标准物质(富铅锌矿石)GBW07165(推荐值As 1 500 μg/g,Hg 114 μg/g)于12支消解管内,每3支为一组,分别加入4种不同消解液,按表1设定步骤进行消解,测量体积为100 mL。使用不同消解试剂测量结果见表3。

表3 使用不同消解液的测试结果
由表3可以看出,只有单独使用HNO3消解时,溶液混浊,相对误差较大,其余消解液测量结果均满意,但考虑氢氟酸在赶酸仪内不能完全赶尽,残留的氢氟酸会给玻璃器皿和仪器带来损害,因此尽量不选用;高氯酸是一种强氧化剂和脱水剂,热、浓高氯酸遇到有机物常发生爆炸,因藏药原材料中含有大量有机物,即便使用硝酸进行了预消解,依然会有残留,使用高氯酸存有潜在危险,因此也不宜选用。综合考虑以上因素,本实验选择8 mL HNO3+H2O2(4+1,V/V)作为消解试剂。
2.3 消解温度和时间的选择
取15支消解管,分别加入ρ(As)=50 μg/mL砷标准溶液和ρ(Hg)=10 μg/mL汞标准溶液各1 mL,再准确称取0.100 0 g标准物质(GBW07165)于消解管内,加入5 mL HNO3+H2O2(4+1,V/V)消解液,分别在不同温度和时间下进行消解,测量体积为100 mL,测得浓度及回收率见表4。
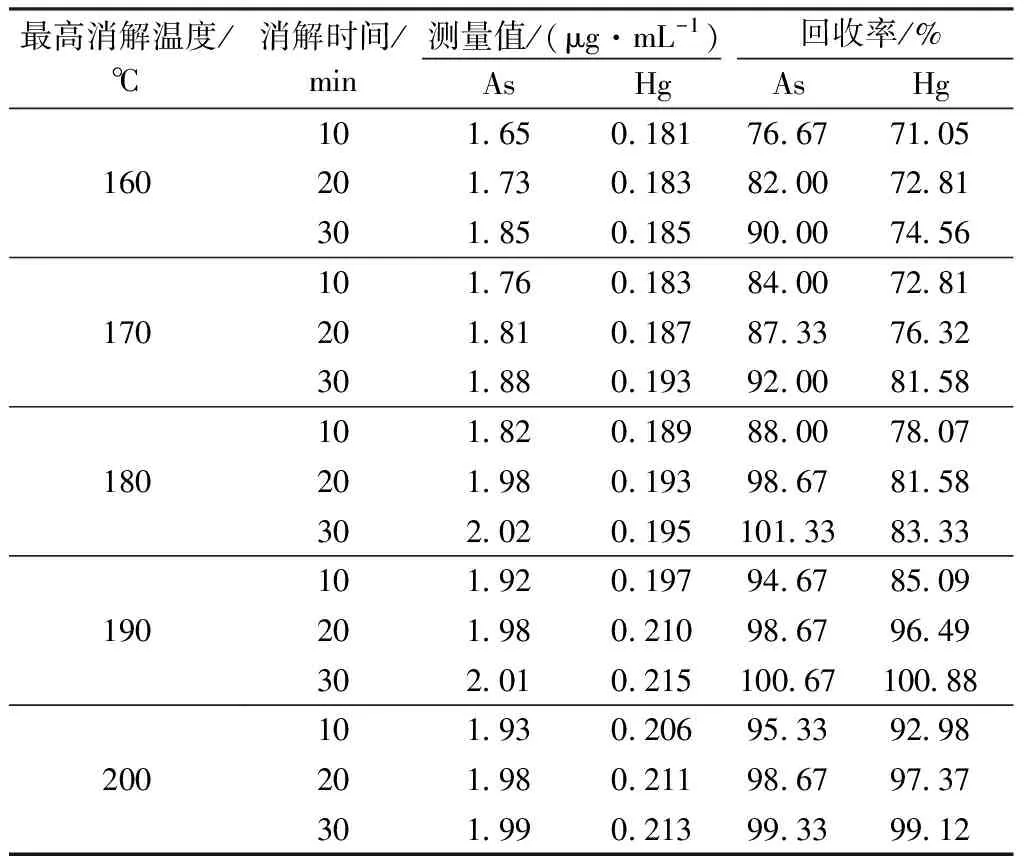
表4 不同消解温度与时间下测定的回收率
从表4中可以看出,砷元素在180 ℃消解30 min时,回收率达到最高,190 ℃变化不明显;200 ℃以后略有下降;汞元素在190 ℃消解30 min时回收率达到最高,随后的变化不明显。因此,同时测定两种元素时,实验选择消解温度为190 ℃,消解时间为30 min。
2.4 酸度的选择
电感耦合等离子体发射光谱仪对酸度的要求没有那么严格,但酸度过小,溶液不易保存,酸度过大,则会抑制分析信号。实验选择5%、10%、15%、20%、25%、30%酸度的混合标准溶液ρ(As,Hg)=20 μg/mL和ρ(As,Hg)=200 μg/mL进行测定,测定结果见图1、2。
从图1、2可以看出,当酸度大于20%后,测定值呈现明显下降趋势,实验结果与文献[12-13]表述一致,因此,实验选择(10+90,V/V)王水作为样品溶液介质。
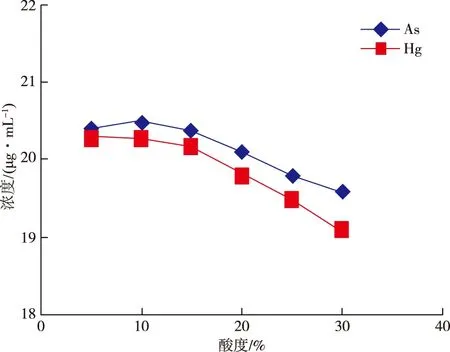
图1 20 μg/mL标准溶液在不同酸度下的测量值Figure 1 As and Hg contents measured in 20 μg/mL standard solution under different acidity.
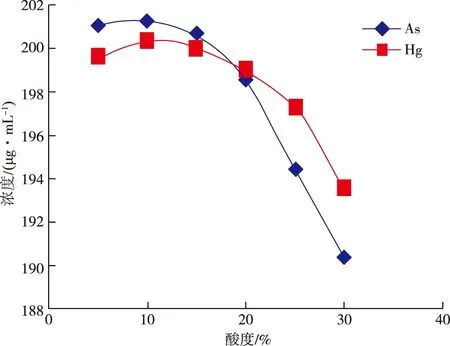
图2 200 μg/mL标准溶液在不同酸度下的测量值Figure 2 As and Hg contents measured in 200 μg/mL standard solution under different acidity.
2.5 选择分析谱线
分析谱线的选择对于电感耦合等离子体发射光谱法的影响是至关重要的,它将直接影响分析结果的正确性。原则上选择灵敏度高而干扰少的谱线。因藏药原材料中砷和汞的含量较高,故而宜选择干扰少、灵敏度较高、线性范围宽且强度适中的谱线作为分析线,经合理扣除背景,而确定最终谱线,见表5。
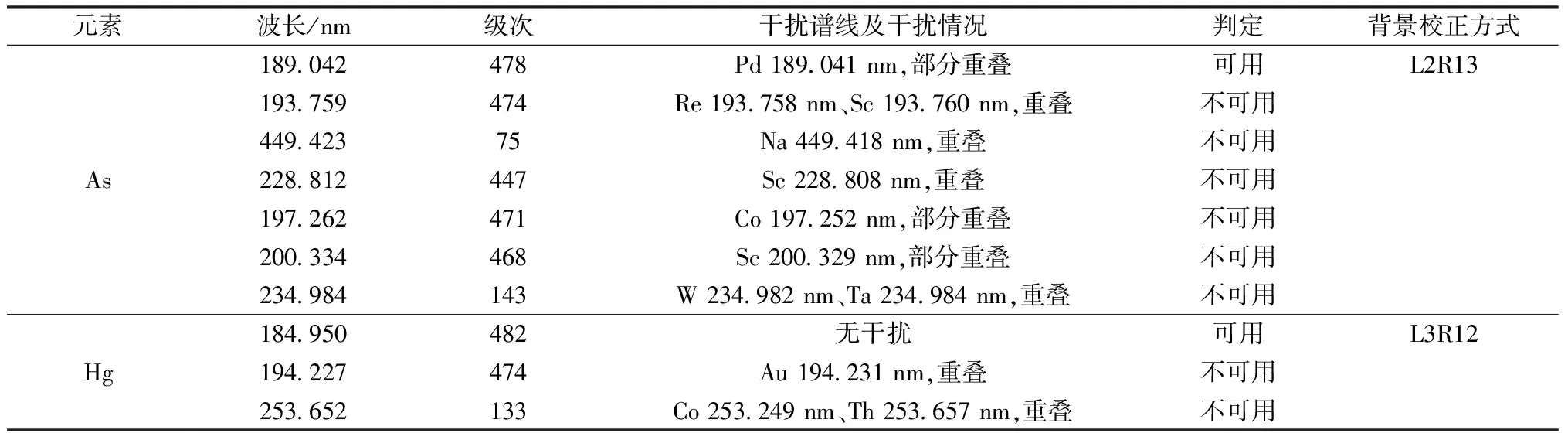
表5 分析谱线及干扰情况
由表5可知,As元素选择189.042{478} nm作为分析谱线,虽然Pd在附近有干扰,但藏药原材料中几乎不含Pd,因此该干扰可忽略不计,而Hg元素则选择184.950{482} nm作为分析谱线。
2.6 校准曲线与检出限
实验按表1设定的工作条件将7个标准系列点进行测定,以砷、汞的质量分数为橫坐标,以发射强度为纵坐标,绘制砷、汞校准曲线(表6)。在该条件下对空白样品连续进行11次平行测量,以测定结果的3倍标准偏差计算方法的检出限,以4倍检出限计算砷、汞的定量限。

表6 校准曲线、检出限和定量限
3 样品分析
3.1 精密度和加标回收实验
按照实验方法测定藏药原材料实际样品中的As、Hg,结果的相对标准偏差(RSD,n=11)As为2.0%,Hg为1.8%,加标回收率为96.0%~103%,见表7。
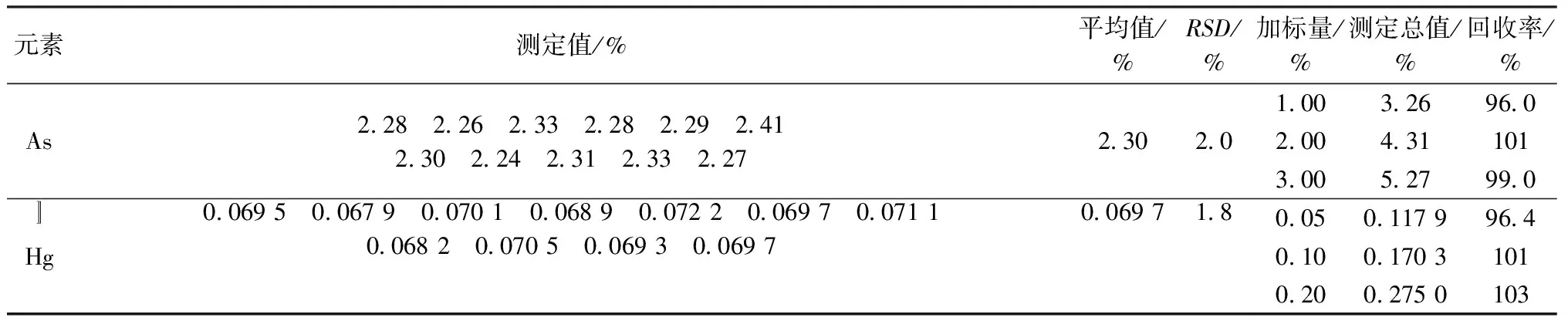
表7 精密度和加标回收实验结果
3.2 方法对比实验
实验选择5个实际样品,分别采用容量法与电感耦合等离子体发射光谱法进行了测试,对比结果见表8。
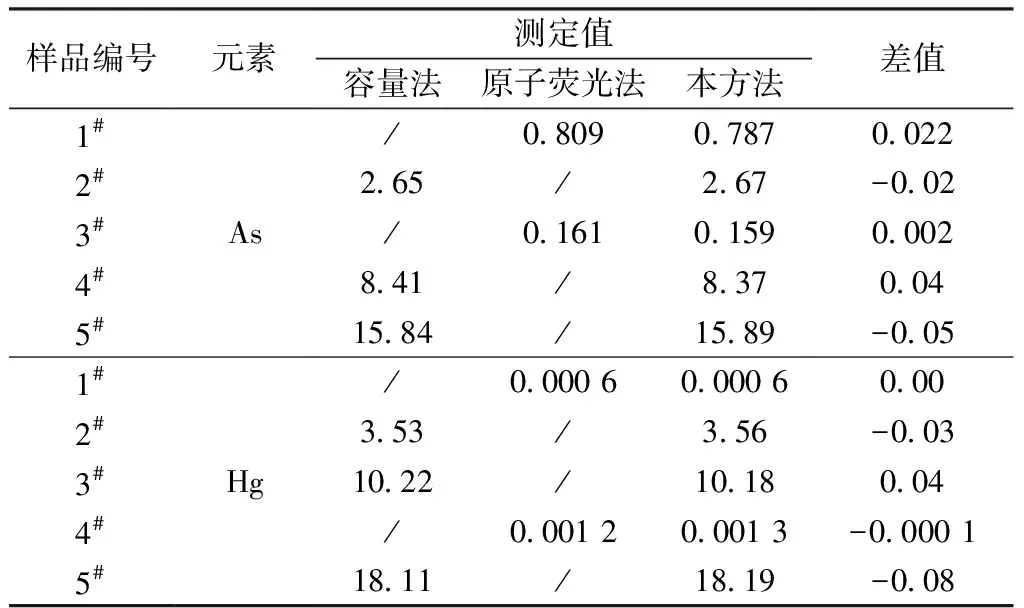
表8 结果对比汇总
从表8数据可以看出,本方法与容量法和原子荧光光谱法的测定结果无明显差异,测定范围更明显优于其他两种方法。
4 结论
藏药原材料成份复杂,因“以毒攻毒”的医理,有毒有害元素占比较高,配比入药时,若不能以准确的测试数据作为指导,或将影响药效,甚至对人身造成伤害。本文采用微波消解法进行样品前处理,并采用电感耦合等离子体发射光谱法进行测定。前处理采用微波密闭消解,大大降低了待测元素挥发的风险,省时省力,同时避免了前处理过程中酸气以及砷、汞等有害元素挥发对实验人员的侵害。该方法具有良好的线性关系,并且具有较低检出限和较高的精密度,加标回收率在96.0%~103%,实验证明,该方法可以快速高效地测定藏药原材料中的砷、汞,为藏药配比提供可靠依据。
