基于HPLC指纹图谱结合化学模式识别及多成分定量的蒲公英质量评价研究
2022-12-28吴晨曦王秀萍李赵嘉
孟 然,吴 哲,冯 薇,吴晨曦,王秀萍*,李赵嘉*
基于HPLC指纹图谱结合化学模式识别及多成分定量的蒲公英质量评价研究
孟 然1, 2,吴 哲1, 2,冯 薇1, 2,吴晨曦3,王秀萍1, 2*,李赵嘉1, 2*
1. 河北省农林科学院滨海农业研究所,河北 唐山 063299 2. 唐山市蒲公英工程技术研发中心,河北 唐山 063299 3. 华北理工大学中医学院,河北 唐山 063210
建立蒲公英的HPLC指纹图谱及多成分含量测定方法,优化指纹图谱测定条件,结合化学模式识别法对不同产地蒲公英进行质量评价,为蒲公英质量控制提供参考。采用Box-Behnken响应面分析法优化蒲公英主要有效成分的制备工艺。采用Mars ODS-AQ色谱柱(250 mm×4.6 mm,5 µm);以甲醇-0.2%磷酸水溶液(35∶65)为流动相,等梯度洗脱;体积流量为1 mL/min;检测波长323 nm;进样量10 μL;柱温30 ℃;检测时间为12 min;对10个不同产地的30批蒲公英建立HPLC指纹图谱,并对5个成分含量进行测定,通过相似度评价、聚类分析及主成分分析对蒲公英质量进行评价。蒲公英主要有效成分的最佳制备工艺为料液比1∶55、甲醇体积分数72%、超声温度80 ℃、超声时间79 min,在此条件下OD值为0.93。建立了蒲公英HPLC指纹图谱,30批蒲公英的相似度在0.647~0.980,标定6个共有峰,指认出5个色谱峰。单咖啡酰酒石酸、绿原酸、咖啡酸、阿魏酸、菊苣酸质量分数分别为0.426%~1.856%、0.007%~0.117%、0.023%~0.101%、0.003%~0.025%、0.311%~1.412%。聚类分析将10个产地的蒲公英分为4类,主成分分析结果表明哈尔滨和沈阳产地的质量较优,并确定单咖啡酰酒石酸、绿原酸、咖啡酸和菊苣酸可作为蒲公英质量评价的主要指标。确定了蒲公英主要有效成分最佳制备工艺,提取率远高于药典方法。建立的蒲公英指纹图谱结合多成分含量测定及化学模式识别法准确、高效、全面地评价蒲公英质量,为蒲公英的质量控制提供依据。
蒲公英;指纹图谱;单咖啡酰酒石酸;绿原酸;咖啡酸;阿魏酸;菊苣酸;化学模式识别;质量评价
蒲公英是菊科植物蒲公英Hand. -Mazz.、碱地蒲公英Kitam.或同属数种植物的干燥全草,主要分布于我国河北、陕西、辽宁和黑龙江等地,临床上具有清热解毒、消肿散结、利尿通淋的作用,常用于治疗疔疮肿毒、乳痈、咽痛、肠痈、湿热黄疸、热淋涩痛等症[1]。现代药理研究表明,蒲公英具有抗氧化、抗炎、抗癌和降血糖等作用[2-3],其中酚酸类化合物是蒲公英的主要有效成分,是发挥药效的重要物质基础。目前从蒲公英中分离得到的酚酸类化合物有32种(表1)[4-7],《中国药典》2020年版以菊苣酸的含量作为蒲公英质量评价指标,仅靠单个主效成分不足以全面、综合地反映蒲公英整体质量。此外,由于蒲公英同属植物多种类、多产地、多成分及相互作用复杂等因素影响,造成不同产地蒲公英质量的差异性。已有文献报道蒲公英饮片、栓剂、配方汤剂等质量研究[8-10],但仍存在研究不系统、分析方法单一的问题,未能较直观地反映出不同批次蒲公英的差异标志物。因此,蒲公英质量综合性评价显得尤为重要。
中药指纹图谱与化学模式识别相结合,能真实、形象地反映中药质量差异,揭示复杂化合物之间的规律,已成为目前最常用最有效的控制天然药材质量的手段[11-12]。本研究建立了蒲公英高效液相色谱(high performance liquid chromatography,HPLC)指纹图谱,同时高效测定单咖啡酰酒石酸、绿原酸、咖啡酸、阿魏酸和菊苣酸5个成分含量,利用响应面法优化蒲公英主效成分的最佳制备方法,结合相似度评价、相关性分析、聚类分析、主成分分析等化学模式识别法对10个不同产地30批蒲公英进行质量分析研究,明确造成产地差异的标记性成分,旨在为蒲公英质量标准的建立提供科学依据。

表1 蒲公英酚酸类化合物
1 仪器与材料
1.1 仪器
Agilent 1200型高效液相色谱仪(美国安捷伦公司);DHY-300型超微粉碎机(北京东华原医疗设备有限责任公司);AP124W型电子天平(日本岛津公司);HH-6型数显恒温水浴锅(上海力辰邦仪器科技有限公司);JT-1027HTD型机械超声波清洗机(深圳市洁拓超声波清洗设备有限公司)。
1.2 材料
单咖啡酰酒石酸(批号SC5580,质量分数≥98.0%)、绿原酸(批号SC8210,质量分数≥98.0%)标准品购自北京索莱宝科技有限公司;咖啡酸(批号YZ-110885,质量分数≥99.7%)、阿魏酸(批号YZ-110773,质量分数≥99.4%)、菊苣酸(批号YZ-111752,质量分数≥98.0%)对照品均购自中国药品生物制品检定所;磷酸、甲醇(色谱纯,美国Fisher Chemical公司);无水甲醇(分析纯,天津市恒兴化学试剂制造有限公司);屈臣氏蒸馏水(广州屈臣氏食品饮料有限公司)。本实验收集了10个产地30批蒲公英,经河北省农林科学院滨海农业研究所王秀萍研究员鉴定均为菊科植物蒲公英Hand. -Mazz,样品信息见表2。
2 方法与结果
2.1 色谱条件
色谱柱为Mars ODS-AQ(250 mm×4.6 mm,5µm);流动相甲醇(A)-流动相0.2%磷酸水溶液(B)(35∶65),等梯度洗脱,体积流量1 mL/min,进样量10 μL,柱温30 ℃,检测波长323 nm,检测时间12 min。
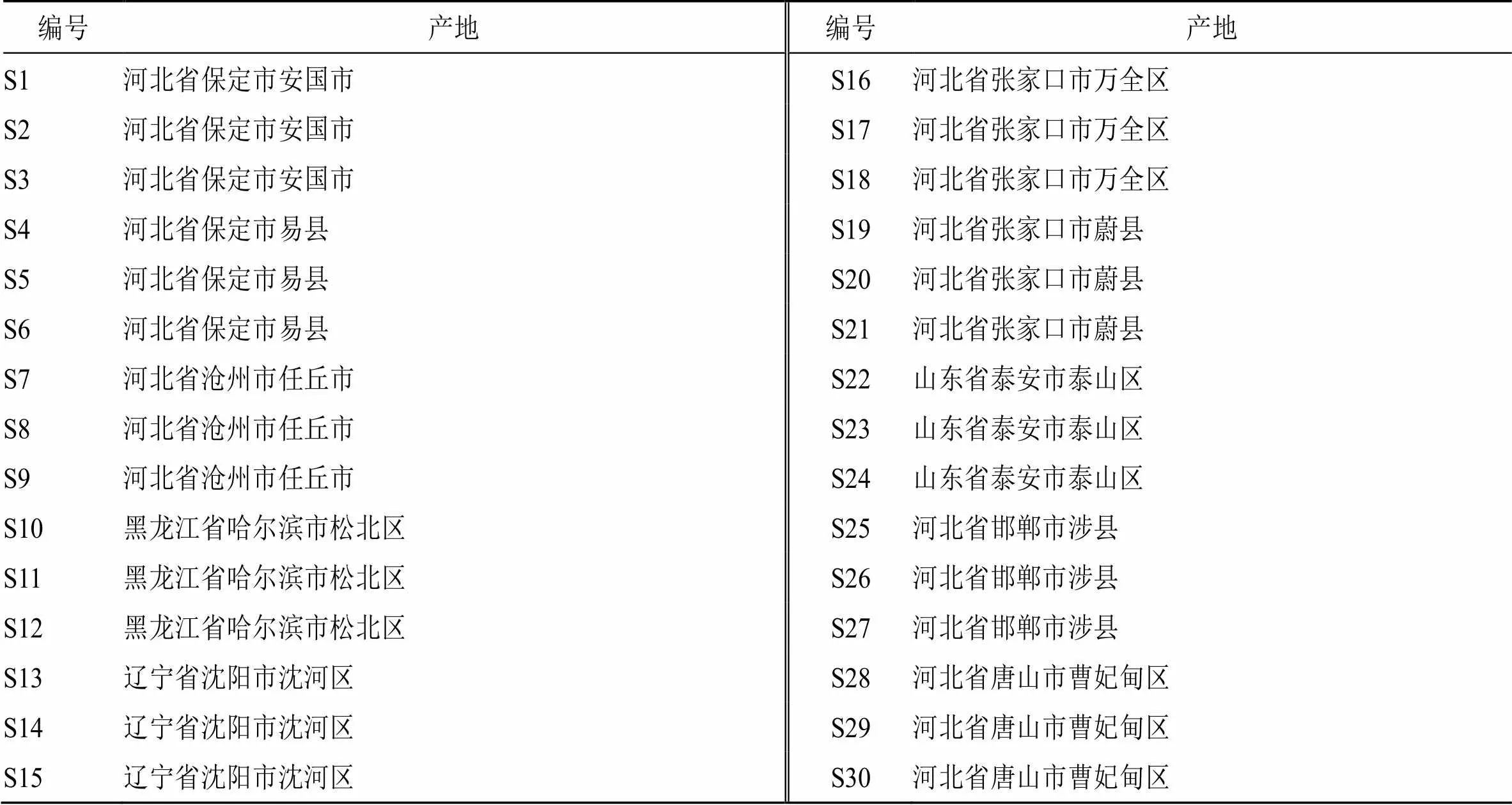
表2 蒲公英药材信息
2.2 溶液的制备
2.2.1 供试品溶液的制备 精密称取蒲公英样品粉末(过四号筛)0.50 g,置于具塞锥形瓶中,精密加入80%甲醇20 mL,称定质量,超声处理(功率400 W,频率40 kHz)20 min,放冷,再称定质量,用80%甲醇补足减失的质量,摇匀,用0.45 µm的针式过滤器滤过,取续滤液,即得供试品溶液。
2.2.2 混合对照品溶液的制备 精密称取单咖啡酰酒石酸、绿原酸、咖啡酸、阿魏酸、菊苣酸对照品适量,分别置10 mL量瓶中,加80%甲醇溶液,超声溶解,定容,制成质量浓度分别为0.975、0.260、0.156、0.126、1.125 mg/mL的对照品贮备液。然后分别精密量取各对照品贮备液2.00 mL,置同一10 mL量瓶中,加80%甲醇定容,摇匀,制得单咖啡酰酒石酸为0.195 mg/mL、绿原酸为0.052 mg/mL、咖啡酸为0.031 mg/mL、阿魏酸为0.025 mg/mL、菊苣酸为0.225 mg/mL的混合对照品溶液。
2.3 方法学考察
2.3.1 线性关系考察 精密吸取“2.2.2”项下混合对照品溶液1.00、2.00、3.00、4.00、5.00、6.00 mL,分别置10 mL量瓶中,加80%甲醇溶液至刻度,摇匀,作为系列对照品溶液,在“2.1”项色谱条件下进样分析,以各组分峰面积为纵坐标(),以对照品溶液的浓度为横坐标(),进行线性回归。结果见表3。
2.3.2 精密度试验 精密吸取“2.2.2”项下方法制备的蒲公英供试品(S7)20 μL,按“2.1”项色谱条件下重复进样6次,记录色谱图。以单咖啡酰酒石酸(1号峰)为参照峰,考察各共有峰的相对保留时间与相对峰面积。结果表明各共有峰相对保留时间和相对峰面积RSD值均小于1.54%,表明仪器精密度良好。

表3 蒲公英中主效成分的线性关系考察结果
2.3.3 重复性试验 精密称取同一批蒲公英供试品(S7)6份,按“2.2.1”项下方法制备供试品溶液,在上述色谱条件下测定,以单咖啡酰酒石酸(1号峰)为参照峰,考察各共有峰的相对保留时间与相对峰面积。结果表明各共有峰相对保留时间和相对峰面积RSD值均小于2.06%,表明方法重复性良好。
2.3.4 稳定性试验 精密称取同一批蒲公英供试品(S7),按“2.2.1”项下方法制备供试品溶液,分别在 0、2、4、6、8、10、12 h时按上述色谱条件进样分析,以单咖啡酰酒石酸(1号峰)为参照峰,考察各共有峰的相对保留时间与相对峰面积。结果表明各共有峰相对保留时间和相对峰面积RSD值均小于2.17%,表明供试品溶液在12 h内稳定性良好。
2.4 蒲公英指纹图谱的建立及相似度评价
2.4.1 蒲公英指纹图谱的建立及共有峰的标定 将30批蒲公英样品按“2.2.1”项下优化的提取工艺条件制备供试品溶液,按“2.1”项下色谱条件进行色谱分析,记录色谱图。将所得的色谱图导入“中药色谱指纹图谱相似度评价系统(2012版本)”软件进行评价,设定S10样品图谱作为参照图谱,时间窗宽度为0.1 min,采用中位数法结合多点校正,进行色谱峰匹配,生成对照指纹图谱。选择蒲公英中的主要成分且分离较好的色谱峰作为共有峰,共标定了6个共有峰。通过与对照品比对,指认出其中5个色谱峰,分别为单咖啡酰酒石酸(峰1)、绿原酸(峰2)、咖啡酸(峰3)、阿魏酸(峰4)和菊苣酸(峰5)。30批蒲公英HPLC指纹图谱见图1,蒲公英对照指纹图谱见图2。
2.4.2 蒲公英指纹图谱的相似度评价 将30批蒲公英指纹图谱与对照图谱进行相似度评价,相似度结果如表4所示,30批蒲公英指纹图谱的相似度在0.647~0.980,相似度在0.9以上的蒲公英占70.00%;30批蒲公英各共有峰相对保留时间的RSD均小于2.02%,相对峰面积的RSD为7.69%~67.75%,相差较大,表明各批次间这6个共有峰所代表化合物的含量存在较大差异,不同产地间蒲公英质量参差不齐。
2.5 蒲公英提取工艺优化
由蒲公英的指纹图谱可知检测的蒲公英色谱峰的峰面积较小,所采用的制备方法提取率不高。因此,本研究对指纹图谱中指认的单咖啡酰酒石酸、绿原酸、咖啡酸、阿魏酸、菊苣酸5个主要有效成分的制备工艺进行优化,进一步对30批不同产地蒲公英进行含量测定。

图1 30批蒲公英的HPLC指纹图谱
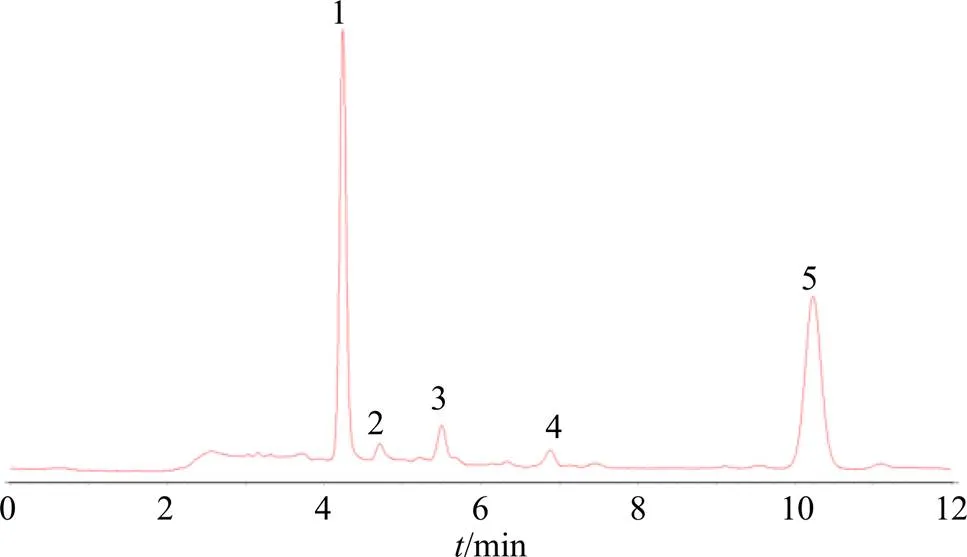
1-单咖啡酰酒石酸 2-绿原酸 3-咖啡酸 4-阿魏酸 5-菊苣酸
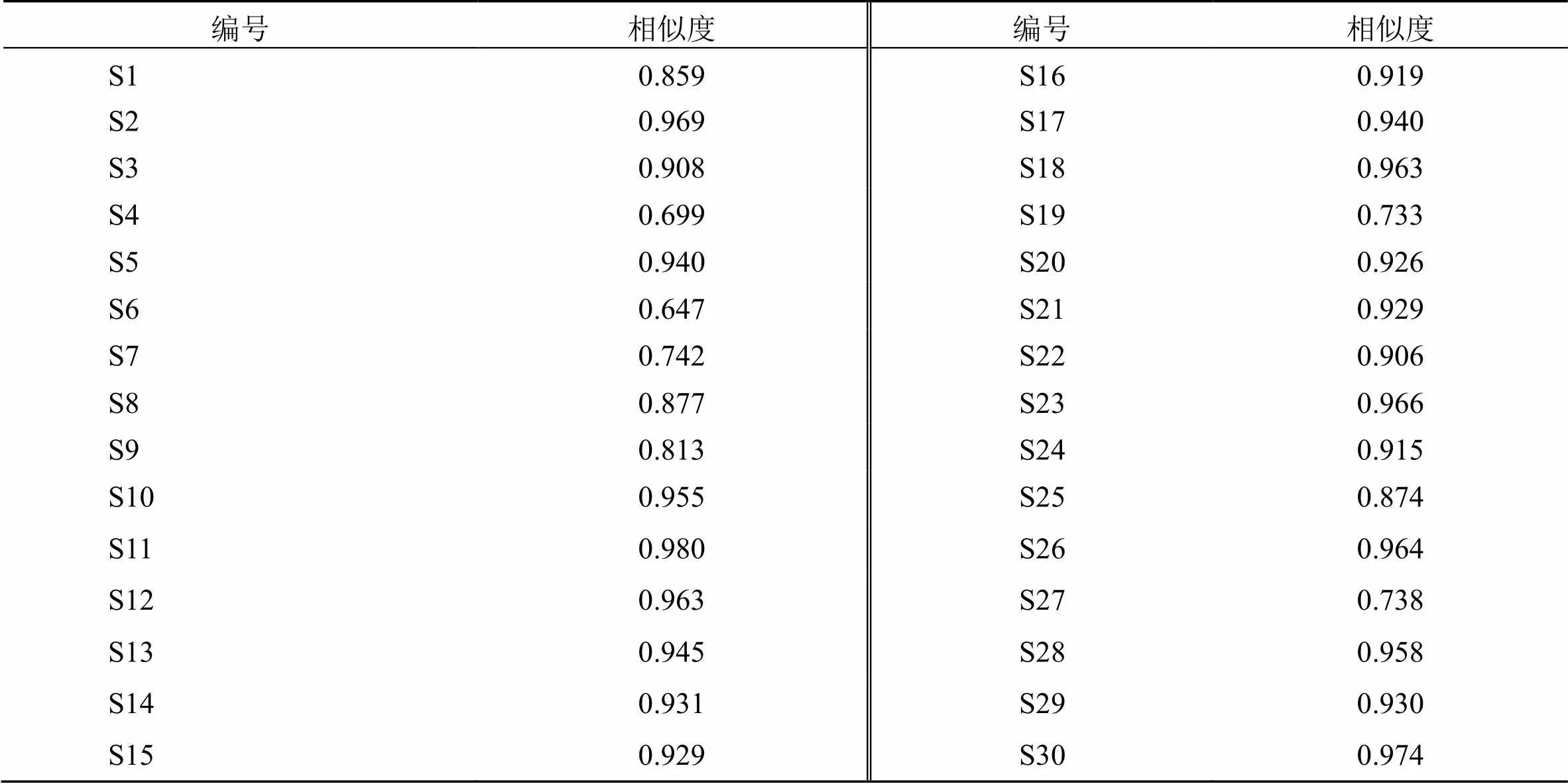
表4 30批蒲公英相似度评价结果
2.5.1 响应面法优化提取工艺试验设计与结果 采用Design-Expert 8.0.6软件,根据Box-Behnken中心组合试验设计原理,结合单因素实验结果,对蒲公英提取工艺进一步优化。为将5个指标成分的效应值综合成一个可以反映总体效应结果的数值,数据处理采用“总评归一化法”[13],对于综合数值越大越好的因素,总评归一值(overall desirability,OD)计算公式如下:
d=(Y−min)/(max−min)
OD=(1×2……×)1/n
为考察指标总数,d为各指标值。
以料液比(A)、甲醇体积分数(B)、超声温度(C)和超声时间(D)作为自变量,以单咖啡酰酒石酸(1)、绿原酸(2)、咖啡酸(3)、阿魏酸(4)和菊苣酸(5)含量的OD值为因变量,设计4因素3水平的响应面优化实验,因素水平设定见表5。
蒲公英主效成分含量=(×10−6××)/×100。
为提取液各成分的质量浓度;为粗提液体积;为稀释倍数;为原料质量。

表5 因素水平设定
根据因素水平设定表,按照“2.2.1”项下方法制备供试品溶液,在“2.1”项色谱条件下测定单咖啡酰酒石酸、绿原酸、咖啡酸、阿魏酸和菊苣酸的含量。Box-Behnken优化实验设计及结果见表6。

表6 Box-Behnken优化实验设计及结果
2.5.2 模型的建立与显著性检验 应用Design-Expert进行回归拟合分析,可得到OD值对料液比(A)、甲醇体积分数(B)、超声温度(C)和超声时间(D)二次多项回归方程:OD值=0.99+0.25 A+0.11 B+0.18 C−0.051 D+0.19 AB+0.18 AC+0.028 AD+0.13 BC−0.095 BD+0.084 CD−0.35 A2−0.30 B2−0.17 C2−0.33 D2;方差分析结果见表7,模拟项<0.01,极显著,表明本试验所选用的二次多项模型具有高度的显著性;失拟项=0.091 7,不显著,说明建立模型不存在失拟因素,拟合程度好[14];R=0.854 1,2adj=0.806 6,表明此模型拟合度较好。因此,该模型能够预测各因素对蒲公英提取含量的影响。因素A、A2、B2、D2达到极显著水平(<0.01),说明对蒲公英主效成分含量有极显著的影响。由值可知,4个因素对蒲公英主效成分提取含量的影响依次是A>C>B>D。
响应面曲线梯度反映各因素对主效成分提取含量的影响大小,响应曲面越陡说明该因素的影响越显著[15]。根据图3曲线上各因素的坡度变化及等高线疏密可知,料液比对主效成分提取含量的影响最大,且因素之间存在一定的交互作用,其中A、B2因素间交互作用显著(<0.05)。各因素对提取含量的影响由大到小的顺序为:A>C>B>D,这与方差分析结果一致。

表7 回归方程系数显著性检验
*表示差异显著(<0.05),**表示差异极显著(<0.01)
*indicates significant difference (< 0.05),**indicates extremely significant difference (< 0.01)

图3 各因素交互作用对OD值影响的响应面图
2.5.3 最佳条件的预测及验证试验 通过回归模型的预测,得到超声波辅助提取蒲公英主效成分的最佳制备工艺为:料液比1∶55.27、甲醇体积分数71.63%、超声温度80.00 ℃、超声时间79.49 min,此时OD值最大为0.93。结合生产实际,将各因素进行调整为:料液比1∶55、甲醇体积分数72%、超声温度80 ℃、超声时间79 min。为了检验方法预测的结果,用试验中得到的最佳制备工艺条件重复试验,平行3份。结果表明,在此条件下进行3次平行实验进行验证,平均OD值为(0.928±0.005),与理论预测值误差值小于0.689%,且经检验的结果差异不显著(>0.05),表明建立的工艺模型具有较好的预测性,可运用此方法制备蒲公英主效成分。
2.5.4 蒲公英主效成分含量测定 取30批蒲公英样品,按照“2.2.1”项下优化的工艺条件制备供试品溶液,按“2.1”项下色谱条件测定,以外标法计算样品中5种主效成分的含量,结果见表8,30批蒲公英中5个主效成分含量有所差异,单咖啡酰酒石酸含量为0.426%~1.856%,绿原酸含量为0.007%~0.117%,咖啡酸含量为0.023%~0.101%,阿魏酸0.003%~0.025%。S11单咖啡酰酒石酸、菊苣酸含量最高,S27中绿原酸、咖啡酸含量最高,S30中阿魏酸含量最高。
在上述5种蒲公英主效成分中,《中国药典》2020年版只对菊苣酸进行了限定,要求含量不得低于0.45%[1],本研究也以此作为菊苣酸的限量标准。测定结果显示,30批蒲公英中菊苣酸的含量为0.311%~1.412%,各产地蒲公英的菊苣酸含量差异明显,绝大部分产地的蒲公英菊苣酸含量达标,其中黑龙江哈尔滨、河北唐山产地的蒲公英菊苣酸含量远远高出药典规定。
2.6 化学模式识别分析
2.6.1 相关性分析 蒲公英成分指标间存在相互依存和制约的关系,多样本2指标之间系数绝对值越大,则这两指标之间的联系越紧密[16]。利用SPSS 26.0软件双变量Pearson相关分析法对5个主效成分之间的相关性进行分析,由表9可知,除酒石酸与绿原酸之间相关性不显著(>0.05)、酒石酸与阿魏酸呈显著正相关(<0.05)之外,其他成分指标之间均呈现两两极显著正相关。酒石酸与咖啡酸、菊苣酸均呈极显著正相关(<0.01),与阿魏酸呈显著正相关(<0.05),其中酒石酸与菊苣酸之间相关性较高,相关系数为0.687;绿原酸与咖啡酸、阿魏酸、菊苣酸均呈极显著正相关(<0.01),其中绿原酸与咖啡酸之间相关系数达0.699,说明两者间关系紧密;咖啡酸与阿魏酸、菊苣酸呈极显著正相关(<0.01);阿魏酸与菊苣酸呈极显著正相关(<0.01);结果表明酒石酸与绿原酸之间相互影响不显著,其他主效成分含量之间相互显著影响,进而影响蒲公英的质量。

表8 30批蒲公英5种主效成分含量测定结果(n=3)
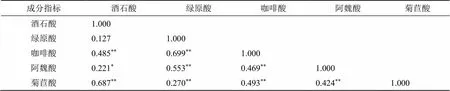
表9 30批蒲公英5种主效成分的相关性分析
*表示水平上(双侧)显著相关(<0.05),**表示水平上(双侧)极显著相关(<0.01)
*indicates a significant correlation (< 0.05) on the level (two-sided), **indicates a extremely significant correlation (< 0.01) on the level (two-sided)
2.6.2 聚类分析 利用SPSS 26.0软件,以30批蒲公英中的酒石酸、绿原酸、咖啡酸、阿魏酸和菊苣酸含量为变量,采用组间联接的聚类方法,以平方欧式距离为样品间距离进行聚类分析,构建树系图(图4),将样本划分为不同类群进行评价。由图可知,各批次蒲公英样本主要分为2大类、4小类:S10~S15聚为一大类(II),其他样本聚为一大类(I)。在平方欧式距离8处,S1~S9、S16~S21、S25~S30聚为一小类(Ia),S22~S24聚为一小类(Ib);S10、S11聚为一小类(IIa),S12~S15聚为一小类(IIb)。从总体数据来看,不同产地蒲公英质量具有一定差异,其中第II类与其他样本差异较大,距离为25时就可以归为不同的类型,这说明黑龙江哈尔滨和辽宁沈阳产区的蒲公英与其他产地蒲公英具有明显的差异;在距离为1时所有样本完全分离,说明虽然30批蒲公英样本来自于不同产地,但仍具有较强相似性。
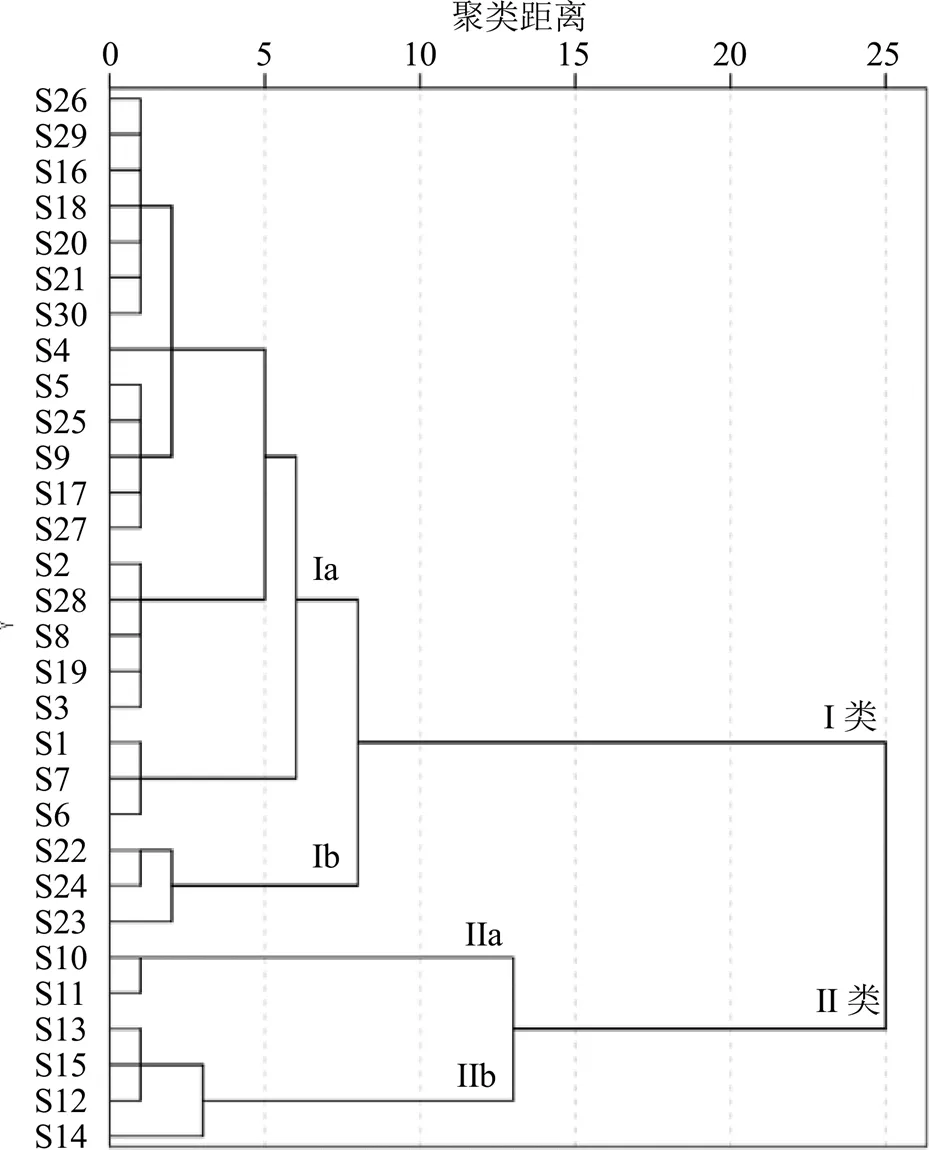
图4 30批蒲公英聚类分析树状图
2.6.3 主成分分析 为了进一步比较不同产地蒲公英样品的质量差异,以30批蒲公英中的酒石酸、绿原酸、咖啡酸、阿魏酸和菊苣酸含量为变量,采用SPSS 26.0软件进行主成分分析,得出主成分的特征值、贡献率、主成分载荷矩阵及特征向量矩阵等。选取其中特征值大于1的前2个因子作为主成分1与主成分2,贡献率分别为59.558%和21.596%,累积贡献率为81.155%(表10),故2个主成分可以基本反映本实验测定的所有成分指标的信息。载荷值反映了各品质指标对主成分矩阵中的权重,数值绝对值的大小反映了原始变量对于因子影响的强度,正负反映了方向[17]。表11显示了主成分分析载荷值的分布,酒石酸、绿原酸、阿魏酸和咖啡酸在第1主成分上载荷较大,表明与PC1相关系数较高,且均成正相关,第1主成分主要反映了这些元素的信息;菊苣酸在第2主成分上载荷较大,即与PC2相关系数较高,表明第2主成分主要反映了菊苣酸的信息。

表10 特征值和累积方差贡献率

表11 主成分载荷矩阵
根据公式=×计算各主成分值,其中原始数据标准化得标准化矩阵,为标准化的正交特征向量矩阵。根据各主成分的贡献率,计算出综合得分,其公式为综=0.595 6×F+0.216 0×F。各主成分值及综合排序结果见表12,根据得分情况反映各产地蒲公英的质量情况,总得分越高表明该地区蒲公英的综合质量越好,本研究中S10~S15综合得分较高,说明来自哈尔滨和沈阳的蒲公英质量相对较优。分别以PC1与PC2建立坐标系,得到主成分分析散点平图(图5),由图可知,PC1和PC2可将样品分为4类,第1类为S10、S11,第2类为S12~S15,第3类为S22~S24,其他归为第4类。主成分分析与聚类分析结果相一致,进一步证明了PC1和PC2可代表蒲公英的质量用于分类。
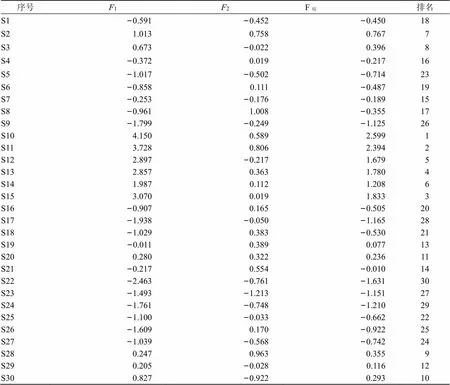
表12 主成分综合得分和排序
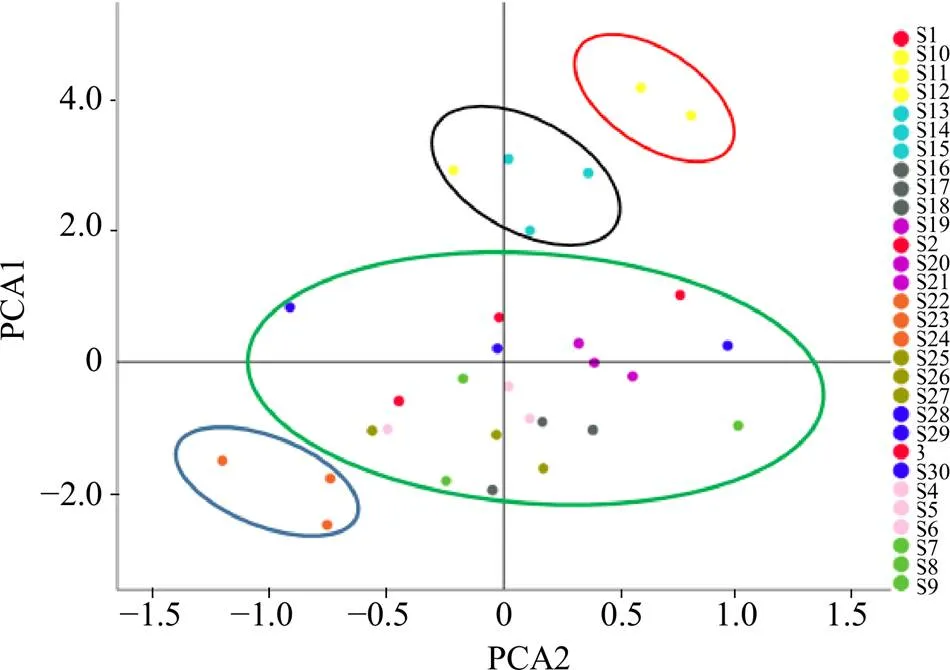
图5 主成分分析散点图
3 讨论
本实验采用《中国药典》2020年版[1]的提取方法,检测得到蒲公英中单咖啡酰酒石酸含量为0.59%,绿原酸含量为0.12%,咖啡酸含量为0.15%,阿魏酸含量为0.08%,菊苣酸为0.363%,计算得OD值为0.09,表明药典提供的方法检测结果可能偏低。因此,本研究以液料比、甲醇浓度、超声时间及温度为因子,采用响应面优化设计,对指纹图谱测定条件进行了优化,得到最佳制备方法为料液比1∶55、甲醇浓度72%、超声温度80.00 ℃、超声时间79 min,此时OD值最大为0.93,大大提高了蒲公英主效成分的提取率。
经查阅文献报道[8,19-20]可知,采用高效液相色谱法检测蒲公英菊苣酸等成分的检测时间通常在26~35 min。在此基础上,本研究对色谱条件进行了系统考察,包括不同流动相系统(乙腈-0.1%甲酸水溶液、甲醇-0.2%磷酸水溶液和甲醇-0.1%甲酸水溶液)、流动相比例(50∶50、45∶55、35∶65)、不同体积流量(0.8、1.0、1.2 mL/min)、不同柱温(25、30、35 ℃)的考察,最终确定了蒲公英分析的最佳色谱条件。在此色谱条件下可将蒲公英中单咖啡酰酒石酸、绿原酸、咖啡酸、阿魏酸、菊苣酸5种主效成分在12 min内检出,从而大大降低了检测时间成本。
产地是影响药材质量的主要原因之一,地形地貌、土壤、气候等因素都可能造成不同产地间的成分差异,同一产地的药材质量也会受采收时间、种植方式等因素影响[18]。本实验通过建立蒲公英HPLC指纹图谱,对10个不同产地30批蒲公英指纹图谱相似度分析,结果显示21批蒲公英样品相似度高于0.9,化学成分组成较为相似。有5批样品相似度低于0.8,可能与生长年限、地理环境条件及贮藏等因素有关,需进一步分析。后续通过方法学考察确定多成分含量测定方法可行,进一步对单咖啡酰酒石酸、绿原酸、咖啡酸、阿魏酸、菊苣酸5种有效成分的含量进行测定,结果发现单咖啡酰酒石酸和菊苣酸含量相对较高,不同产地间含量存在较大差异。
聚类分析中,在平方欧式距离25处,将黑龙江和辽宁的蒲公英聚为一类,而河北7个地区和山东的蒲公英聚为一类。当距离刻度为8时,又将河北7个地区与山东的蒲公英区分开,说明河北7个地区与山东的蒲公英药材质量具有相似性,有可能是因为河北与山东产地相邻、生态环境相似而聚为一类,但也可能因为地处沿海与内陆的区别具有一定差异性;而不能将黑龙江和辽宁2产地的药材清晰地分离出来,说明东北2地的蒲公英药材质量较为接近,可能原因是两个产区同属于东北地区,药材的化学成分很相似,因此造成这2类数据差异性小。
主成分分析结果分析发现酒石酸、绿原酸、阿魏酸和咖啡酸最能代表第1主成分,但阿魏酸峰面积较小;菊苣酸最能代表第2主成分。同时,文献表明,单咖啡酰酒石酸、绿原酸、咖啡酸和菊苣酸为蒲公英主要活性成分—酚酸类物质中重要成分[21],选择酒石酸、绿原酸、咖啡酸和菊苣酸作为药材含量测定指标可以更加全面反映蒲公英内在品质。主成分分析将30批蒲公英样本分为4类,结果与聚类分析结果相一致。根据综合得分可知,来自哈尔滨和沈阳产地的蒲公英质量相对较优。
综上所述,相似度分析、聚类分析、主成分分析是中药指纹图谱结合化学模式识别手段应用最普遍的分析方法,清晰地表明了不同产地蒲公英药材的差异。本研究建立指纹图谱与成分测定、化学模式识别法相结合,能较为科学、全面地反映药材质量。本实验建立的方法操作简便、高效稳定,对蒲公英质量标准的提升具有重要意义。
利益冲突 所有作者均声明不存在利益冲突
[1] 中国药典 [S]. 一部. 2020: 367-368.
[2] Montes F O, A, Fenton-Navarro B. Active compounds of medicinal plants, mechanism for antioxidant and beneficial effects [J]., 2019, 88(1): 1-10.
[3] Faria T C, Nascimento C C H C, De Vasconcelos S D D,. Literature review on the biological effects ofplant in therapy [J]., 2019, 7(3): 94-99.
[4] Grauso L, Emrick S, Bonanomi G,. Metabolomics of the alimurgic plants,andby combined NMR and GC-MS analysis [J]., 2019, 30(5): 535-546.
[5] 聂文佳, 徐帅师, 张咏梅. 蒲公英有效成分及其药理作用研究进展 [J]. 辽宁中医药大学学报, 2020, 22(7): 140-145.
[6] 施树云, 周长新, 徐艳, 等. 蒙古蒲公英的化学成分研究[J]. 中国中药杂志, 2008, (10): 1147-1157.
[7] Jędrejek D, Kontek B, LIS B,. Evaluation of antioxidant activity of phenolic fractions from the leaves and petals of dandelion in human plasma treated with H2O2and H2O2/Fe [J]., 2017, 262: 29-37.
[8] 徐帅师, 聂文佳, 张咏梅. 蒲公英栓剂的制备及质量检查 [J]. 沈阳药科大学学报, 2021, 38(7): 744-750.
[9] 李超, 苏晓楠, 朱法根, 等. 蒲地蓝消炎口服液源头把控: 基于超高效液相指纹图谱的蒲公英药材质量研究 [J]. 中国中药杂志, 2020, 45(18): 4307-4315.
[10] 孙博, 常阿倩, 朱广伟, 等. 基于传统煎药工艺的川牛膝饮片标准汤剂制备及质量评价方法研究 [J]. 世界中医药, 2019, 14(12): 3144-3149.
[11] 李洋, 陈健, 张越, 等. 基于指纹图谱结合化学模式识别及多成分含量测定的白芍药材质量评价研究 [J]. 中草药, 2022, 53(1): 231-237.
[12] 李妍, 何文媛, 王康宇, 等. 基于HPLC多指标成分测定及指纹图谱多模式识别方法的北细辛质量分析 [J]. 中草药, 2022, 53(1): 238-243.
[13] 李欧, 赵善婷, 李亚迪, 等. 总评“归一值”优选黄芩中黄酮类成分的提取工艺 [J]. 中兽医医药杂志, 2021, 40(3): 17-23.
[14] 杨凯, 马子豪, 李源, 等. Box-Behnken响应面法优化白及多糖/聚乙烯醇湿法纺丝工艺及纤维性能评价 [J]. 中草药, 2020, 51(14): 3645-3654.
[15] 郭宏垚, 李冬, 雷雄, 等. 花椒多酚提取工艺响应面优化及动力学分析 [J]. 食品科学, 2018, 39(2): 247-253.
[16] 孟然, 张晓东, 杨雅华, 等. 基于多元统计分析的曹妃甸区24份盐地碱蓬成分指标综合评价 [J]. 食品工业科技, 2021, 42(15): 78-84.
[17] 卢冉, 王炳智, 田英姿. 不同品种杏仁氨基酸组成分析及综合评价 [J]. 食品科学, 2021, 42(24): 229-235.
[18] 林丽, 李欢欢, 谢辉, 等. 基于HPLC指纹图谱结合化学计量学的旋覆花药材质量评价研究 [J]. 中草药, 2021, 52(6): 1751-1758.
[19] 惠西珂, 李超, 谷巍, 等. 蒲公英质量标准提升及不同产地药用蒲公英质量评价 [J]. 中国药房, 2021, 32(7): 818-824.
[20] 刘爱朋, 郭利霄, 薛紫鲸, 等. 基于指纹图谱和多组分含量测定的蒲公英药材质量控制研究 [J]. 中国中药杂志, 2018, 43(18): 3715-3721.
[21] Xie P J, Huang L X, Zhang C H,. Skin-care effects of dandelion leaf extract and stem extract: Antioxidant properties, tyrosinase inhibitory and molecular docking simulations [J]., 2018, 111: 238-246.
Quality evaluation of dandelion based on HPLC fingerprint combined with chemical pattern recognition and multi-component determination
MENG Ran1, 2, WU Zhe1, 2, FENG Wei1, 2, WU Chen-xi3, WANG Xiu-ping1, 2, LI Zhao-jia1, 2
1. Institute of Coastal Agricultural, Hebei Academy of Agriculture and Forestry Sciences, Tangshan 063299, China 2. Tangshan Dandelion Engineering Technology Research Center, Tangshan 063299, China 3. School of Traditional Chinese Medicine, North China University of Science and Technology, Tangshan 063210, China
To establish the HPLC fingerprint and the determination method of multi-component content of dandelion, optimize the conditions of fingerprint determination, and combine the method of chemical pattern recognition to evaluate the quality of dandelion from different origins, so as to provide reference for the quality control of dandelion.The preparation process of main ingredients in dandelion were optimized by Box-Behnken response surface methodology. Mars ODS-AQ column (250 mm×4.6 mm, 5 µm) was used; methanol-0.2% phosphoric acid aqueous solution (35∶65) was used as the mobile phase for gradient elution; the flow rate was 1 mL/min, the detection wavelength was 323 nm; the injection volume was 10 μL; the column temperature was 30 ℃; the detection time was 12 min. HPLC fingerprints were established for 30 batches of dandelion from 10 different origins, and the contents of five components were determined. The quality of dandelion was evaluated by similarity evaluation, cluster analysis and principal component analysis.The optimal preparation process of dandelion was as follows: solid-liquid ratio was 1∶55, methanol concentration was 72%, ultrasonic temperature was 80 ℃, ultrasonic time was 79 min, and the OD value under these conditions was 0.93. The HPLC fingerprint of dandelion was established, the similarity of 30 batches of dandelion was 0.699—0.980, six common peaks were calibrated, and five chromatographic peaks were identified. The mass fractions of caftaric acid, chlorogenic acid, caffeic acid, ferulic acid and cichoric acid were 0.426%—1.856%, 0.007%—0.117%, 0.023%—0.101%, 0.003%—0.025% and 0.311%—1.412%, respectively. The dandelion from 10 origins were divided into four categories by cluster analysis. The results of principal component analysis showed that the quality of Harbin and Shenyang origins were better, and it was determined that caftaric acid, chlorogenic acid, caffeic acid and cichoric acid could be used as the main indicators for the quality evaluation of dandelion.The optimal preparation process for main ingredients of dandelion was determined, and the extraction rate was much higher than that of the pharmacopoeia method. The established dandelion fingerprint, combined with the determination of multi-component content and method of chemical pattern recognition, can evaluate the quality of dandelion accurately, efficiently and comprehensively, and provide a basis for the quality control of dandelion.
dandelion; fingerprint; caftaric acid; chlorogenic acid; caffeic acid; ferulic acid; cichoric acid; chemical pattern recognition; quality evaluation
R286.2
A
0253 - 2670(2022)24 - 7887 - 11
10.7501/j.issn.0253-2670.2022.24.026
2022-06-09
河北省重点研发计划项目(21326312D-8);河北省农林科学院现代农业科技创新工程课题资助(2022KJCXZX-BHS-4);河北省农林科学院基本科研业务费项目资助(2021010101)
孟 然(1993—),女,硕士,助理研究员,研究方向为中草药功能成分提取与分析。E-mail: yoki_meng@163.com
王秀萍,女,研究员,从事中药材鉴定研究。E-mail: bhswxp@163.com
李赵嘉,男,助理研究员,从事中草药高效利用研究。E-mail: tofriendzhaojia@163.com
[责任编辑 时圣明]
