橙皮苷磷脂复合物纳米混悬剂的制备、表征及口服药动学研究
2022-12-28张文周郝海军
李 茜,张文周*,郝海军
橙皮苷磷脂复合物纳米混悬剂的制备、表征及口服药动学研究
李 茜1, 2, 3,张文周1, 2, 3*,郝海军4
1. 郑州大学附属肿瘤医院/河南省肿瘤医院 药学部,河南 郑州 450000 2. 河南省肿瘤精准用药及综合评价工程研究中心,河南 郑州 450000 3. 河南省抗肿瘤药物研究医学重点实验室,河南 郑州 450000 4. 上海市中药研究所,上海 201401
制备橙皮苷磷脂复合物(hesperidin phospholipids complex,HD-PC)纳米混悬剂(HD-PC nanosuspensions,HD-PC-NPs),并考察在SD大鼠体内口服药动学行为。将橙皮苷制备成HD-PC,以提高橙皮苷溶解度。采用纳米沉淀-高压均质法制备HD-PC-NPs。在单因素实验基础上,以稳定剂与HD-PC用量比、高压均质压力和均质次数为主要影响因素,粒径、PDI值和ζ电位的总评归一值(OV)作为考察指标,采用Box-Behnken设计-效应面法优化HD-PC-NPs制备工艺,并制备成冻干粉末。采用透射电子显微镜(TEM)观察HD-PC-NPs形态,透析袋法考察药物释放情况。SD大鼠分为橙皮苷混悬液组、HD-PC组和HD-PC-NPs组,HPLC法测定大鼠血浆中的橙皮苷质量浓度,计算主要药动学参数及相对口服吸收生物利用度。HD-PC-NPs的最处方工艺为稳定剂与HD-PC用量比为3.2,均质压力95 MPa,均质次数为10次,制备温度为50 ℃。5%甘露醇制得的冻干粉末外观饱满。HD-PC-NPs呈球形或类球形,平均粒径为(268.62±18.14)nm,PDI为0.122±0.013,ζ电位为(−31.79±1.37)mV。HD-PC-NPs将橙皮苷的溶解度提高至77.06倍,6 h累积释放率达到94.68%。药动学结果显示,HD-PC-NPs达峰时间显著性提前,半衰期(1/2)延长至(5.69±0.82)h,达峰浓度(max)提高至(1 213.96±149.88)ng/mL,相对口服生物利用度提高至3.09倍。HD-PC-NPs可提高橙皮苷溶解度,促进药物体外溶出及体内吸收。
橙皮苷;磷脂复合物;纳米混悬剂;Box-Behnken设计-效应面法;生物利用度;纳米沉淀-高压均质法
橙皮苷又称柑果苷、陈皮苷,主要存在于柑桔属L.植物中,属于黄酮类化合物。国内外研究[1-4]显示,橙皮苷有抗肿瘤、抗炎、抗氧化、抗病毒、降血压、降血糖、提高免疫力等药理活性。橙皮苷在水中溶解度仅为(6.32±0.12)µg/mL,橙皮苷脂溶性也很差[5],在pH 1.2~7.4时油水分配系数波动区间为0.071~1.280,lg值多为负值[6]。容易受到体内各种代谢酶、肠道菌群等影响[7],口服吸收生物利用度仅为3.51%[6]。纳米技术可提高药物溶解度并促进药物吸收[8],目前,已有橙皮苷脂质体、纳米乳、自微乳等报道[9-11],但存在包封率及载药量低、使用大量表面活性剂等问题,临床应用受到一定限制。
磷脂复合物(phospholipids complex,PC)是难溶性药物与磷脂通过某种作用力(如氢键、范德华力等)结合在一起形成的一种物质,可同时改善水溶性及脂溶性,增加吸收[12-16]。但PC的分散性及溶出度较差,可能限制生物利用度提高幅度,且黏性较大,不便于给药[17]。纳米混悬剂(nanosuspensions,NPs)是将难溶性药物与稳定剂通过制剂手段制得的一种“纯”药物纳米制剂[18-22],制备工艺简单可靠。橙皮苷在乙醇等有机溶剂中溶解度很差,故本研究先将橙皮苷制备成橙皮苷磷脂复合物(helicid phospholipids complex,HD-PC)来改善其溶解度,为后续纳米制剂研究奠定基础。采用纳米沉淀结合高压均质法将HD-PC进一步制备成HD-PC纳米混悬剂(HD-PCnanosuspensions,HD-PC-NPs),并进行体内外评价,为橙皮苷纳米制剂研究提供新思路。
1 仪器、试剂及动物
AL204型电子天平,梅特勒托利多仪器公司;Agilent 1100型高效液相色谱仪,美国安捷伦仪器公司;LC-MS-M1系列加热磁力搅拌器,上海力辰仪器科技有限公司;YYJZ-2型高压均质机,上海永延纳米科技有限公司;Bettersize 3000 Plus型粒度仪,丹东百特仪器有限公司;LGJ-10型实验型冻干机,北京松源华兴生物技术有限公司;TD-SM型台式高速离心机,艾莱宝生物技术有限公司;JC-220B型氮吹仪,聚创华业仪器公司;Talos F200X S/TEM型透射电子显微镜(TEM),北京欧波同光学技术有限公司。
对照品橙皮苷(批号110721-202019,质量分数为95.3%)、毛蕊异黄酮葡萄糖苷(批号111213-202015,质量分数为98.2%),国家食品药品检定研究院;橙皮苷原料药,批号20201030,质量分数为97%,北京百灵威生物医药有限公司;大豆磷脂,批号C1256891,上海麦克林生化科技有限公司;聚乙二醇1000维生素E琥珀酸酯(TPGS),批号20191108,西安海斯夫生物有限公司;甘露醇,批号202005,嘉兴南箭生物材料有限责任公司。
SD大鼠,清洁级,雌雄兼用,购自河南省动物实验中心,许可证号为SCXK(豫)2020-0001,实验室饲养至体质量为(240±20)g。所有动物实验遵循郑州大学附属肿瘤医院/河南省肿瘤医院有关实验动物管理和使用的规定,均符合3R原则。
2 方法与结果
2.1 HD-PC的制备[6]
取干燥后的橙皮苷原料药0.5 g和大豆磷脂0.75 g置于烧杯中,加入80 mL四氢呋喃。于45 ℃水浴中磁力搅拌4 h,得澄清溶液。同温度下减压旋转蒸发,即得HD-PC。
2.2 HD-PC-NPs的制备
纳米沉淀-高压均质法制备HD-PC-NPs[18-19]。溶解度实验显示,HD-PC将橙皮苷在无水乙醇中溶解度由(37.16±0.19)µg/mL提高至(5.69±0.36)mg/mL,为制备HD-PC-NPs奠定基础。取50 mg的HD-PC超声溶于6 mL无水乙醇,得有机相;取一定量的稳定剂溶于50 mL蒸馏水,得水相。在磁力搅拌速度为1000 r/min、温度为50 ℃条件下将有机相缓慢滴至水相,超声5 min后减压旋蒸20 min。循环均质数次,补加蒸馏水至50 mL,采用0.45 μm微孔滤膜滤过,即得HD-PC-NPs混悬液。
2.3 橙皮苷的含量测定
2.3.1 色谱条件 色谱柱为Angilent SB C18柱(250 mm×4.6 mm,5 μm);检测波长283 nm;流动相为乙腈-0.5%醋酸水溶液(22∶78);柱温为30 ℃;体积流量为1.0 mL/min。样品的理论塔板数按橙皮苷峰计均不低于8000。
2.3.2 HD-PC及HD-PC-NPs供试品溶液的配制 取HD-PC约10 mg置于50 mL量瓶中,加30 mL有机溶剂[乙腈-DMSO(9∶1)]超声20 s,放置至室温后加入流动相稀释至刻度,摇匀,过0.45 μm微孔滤膜,即得HD-PC供试品溶液。取HD-PC-NPs混悬液1 mL置于50 mL量瓶中,加30 mL乙腈超声20 s,放置至室温后加入流动相稀释至刻度,摇匀,过0.45 μm微孔滤膜,即得HD-PC-NPs供试品溶液。
2.3.3 对照品溶液的配制 精密称取橙皮苷对照品10 mg,置于250 mL量瓶中,加入4 mL DMSO超声溶解后乙腈稀释至刻度,即得40 μg/mL橙皮苷对照品储备液,密封,置于冰箱中,冷藏备用。
2.3.4 线性关系考察 取橙皮苷对照品储备液,采用乙腈依次稀释至10.00、5.00、1.00、0.50、0.10、0.05 μg/mL的对照品溶液,进样测定。以橙皮苷峰面积为纵坐标(),橙皮苷质量浓度为横坐标()作线性回归,得回归方程=14.296 7-0.659 2,=0.999 8,可见橙皮苷在0.05~10.00 μg/mL具有良好的线性关系。
2.3.5 精密度试验 取橙皮苷质量浓度为10.00、1.00、0.05 μg/mL对照品溶液,分别连续进样6次。结果显示橙皮苷峰面积的RSD依次为0.43%、0.19%、0.52%,可见仪器精密度良好。
2.3.6 稳定性试验 取HD-PC和HD-PC-NPs混悬液供试品溶液,于0、3、6、9、18、24 h时间点进样。结果显示HD-PC和HD-PC-NPs的橙皮苷峰面积的RSD分别为0.81%、1.15%,说明供试品溶液在24 h内稳定性良好。
2.3.7 重复性试验 按照“2.3.2”项下所述方法平行制备6份HD-PC和HD-PC-NPs供试品溶液,分别进样测试橙皮苷的峰面积,计算橙皮苷含量。结果显示,HD-PC和HD-PC-NPs中橙皮苷含量的RSD分别为1.54%和0.79%,说明重复性良好。
2.3.8 加样回收率试验 取9份约含5 mg橙皮苷的HD-PC,分成低、中、高3组,分别按80%、100%、120%的水平加入橙皮苷对照品,按“2.3.2”项下所述方法分别制备供试品溶液。进样测定橙皮苷含量并计算回收率。结果显示,HD-PC平均加样回收率为101.42%,RSD为0.86%。
分别取9份体积为0.5 mL的HD-PC-NPs混悬液,分成低、中、高3组,分别按80%、100%、120%的水平加入橙皮苷对照品,按“2.3.2”同法操作制备供试品溶液后进样测定橙皮苷含量并计算回收率。结果显示,HD-PC-NPs平均加样回收率依次为99.17%,RSD为1.14%,说明准确度较高。
2.4 HD-PC的复合率测定[6]
称10 mg的HD-PC置于25 mL量瓶中,加入氯仿,过0.45 μm微孔滤膜,减压旋转蒸发除去氯仿,加入乙腈复溶,测定参加复合反应的橙皮苷量(1)。按“2.3.2”方法测定橙皮苷总量(0)。计算复合率(复合率=1/0)。结果显示,3批HD-PC的平均复合率为99.91%。
2.5 HD-PC的XRPD研究
取橙皮苷、磷脂、物理混合物(橙皮苷与磷脂比例同HD-PC)和HD-PC适量进行XRPD扫描,采用Cu-Ka靶,扫描速度为8°/min,扫描范围(2)为8°~45°,结果见图1。橙皮苷原料药在8°、10.4°、10.5°、14.7°、14.9°、15.3°、15.8°等处存在特征晶型峰。橙皮苷与磷脂简单混合制得的物理混合物XRPD图中仍可见橙皮苷原料药的晶型峰。而HD-PC的XRPD图谱中未见橙皮苷晶型衍射峰,说明橙皮苷转变为无定型物质,这也证明HD-PC制备成功。
2.6 HD-PC-NPs粒径、多分散系数(PDI)与ζ电位的测定
取HD-PC-NPs混悬液50 μL,加入5 mL蒸馏水混匀,平行制备3份。取适量于粒度分析仪上分别测定HD-PC-NPs粒径、PDI及ζ电位。
2.7 单因素实验
2.7.1 稳定剂种类的考察 固定HD-PC用量为50 mg,稳定剂与HD-PC用量比为3∶1,超声功率为200 W,超声时间为15 min,均质压力为70 mPa,均质次数10次,分别考察不同稳定剂对HD-PC-NPs的影响。结果见表1,采用泊洛沙姆188、PVP K30和聚山梨酯80制得的HD-PC-NPs粒径和PDI值均远大于TPGS(<0.01)。PVP K30和TPGS制得的HD-PC-NPs的ζ电位绝对值较为接近,但两者无显著性差异,故选择最终TPGS作为稳定剂来制备HD-PC-NPs。
2.7.2 HD-PC与TPGS用量比例的考察 固定药物用量50 mg,TPGS作为稳定剂,超声功率为200 W,超声时间为15 min,均质压力70 MPa,均质次数10次,分别考察TPGS与HD-PC不同用量比例对HD-PC-NPs的影响。结果见表2,随着HD-PC用量的增加,HD-PC-NPs粒径和PDI值呈先减小再增大趋势。当TPGS与HD-PC用量比例为3∶1时粒径相对较小,PDI值小于0.3,但ζ电位绝对值仍小于30 mV。因此,后续需要对TPGS与HD-PC用量比例进行优化。

表1 稳定剂种类的影响(, n = 3)
表2 稳定剂与HD-PC用量比例的影响(, n = 3)
2.7.3 超声功率的考察 固定药物用量50 mg,TPGS与HD-PC用量比例为3∶1,超声时间为15 min,均质压力70 MPa,均质次数10次,分别考察不同超声功率对HD-PC-NPs的影响。结果见表3,随着超声功率逐渐增加,粒径和PDI值先减小后增大,提示超声功率可影响粒径大小及均一性。超声功率过大时体系温度较高,可能影响HD-PC稳定性,且粒径和PDI也开始变大,ζ电位绝对值变小。综合HD-PC-NPs的粒径、PDI和ζ电位绝对值,最终选择超声功率为250 W。
表3 超声功率的影响(, n = 3)
2.7.4 超声时间的考察 固定药物用量50 mg,TPGS与HD-PC用量比例为3∶1,超声功率为250 W,均质压力70 mPa,均质次数10次,分别考察不同超声时间对HD-PC-NPs的影响。结果见表4,随着超声时间的增加HD-PC-NPs粒径逐渐变小,但过长的超声时间反而导致粒径变大。由于超声时间15、20 min时HD-PC-NPs的粒径、PDI和ζ电位绝对值无显著性差异(>0.05),为节约能源,故选择超声时间为15 min。
2.7.5 均质压力的筛选 固定药物用量50 mg,TPGS与HD-PC用量比例为3∶1,超声功率为250 W,时间为15 min,均质次数10次,分别考察不同均质压力对HD-PC-NPs的影响。结果见表5,随着均质压力的逐渐增加HD-PC-NPs粒径呈先减小后增大,可能是过高的均质压力促使HD-PC-NPs发生融合,从而使粒径增大。当均质压力达到100 mPa时PDI值仍小于0.2,说明一定的均质压力可使粒径均一化程度提高,但达到140 mPa时PDI值开始增大,且ζ电位绝对值下降。可见,均质压力对粒径、PDI和ζ电位影响较大,故选为主要影响因素。
表4 超声时间的影响(, n = 3)
2.7.6 均质次数的筛选 固定药物用量50 mg,TPGS与HD-PC用量比例为3∶1,超声功率为250 W,时间为15 min,均质压力为100 mPa,分别考察不同均质次数对HD-PC-NPs的影响。结果见表6,增加均质次数可使HD-PC-NPs粒径及PDI值下降,但均质次数过多时粒径及PDI值有增大趋势,且ζ电位绝对值也出现变小情况。可见均质次数对粒径、PDI和ζ电位影响较大,故选为主要影响因素。
2.8 Box-Behnken效应面法优化制备工艺
2.8.1 Box-Behnken试验设计 参考“2.6”项下结果,可见TPGS与HD-PC用量比(1)、高压均质压力(2)及均质次数(3)对HD-PC-NPs各个指标影响较大,故作为主要影响因素。以HD-PC-NPs的粒径、PDI值和ζ电位作为响应值,并将该3个指标转换成总评归一值(overall desirability value,OD),计算过程为①粒径(1)、PDI值(2)和ζ电位(3)计算公式为(max-M)/(max-min);②计算OD值,OD=(12…d)1/k,为指标数。Box-Behnken响应面法试验设计见表7,分别制备不同处方工艺下的HD-PC-NPs,平行3次,结果见表7。
表5 均质压力的影响(, n = 3)
表6 均质次数的影响(, n = 3)
2.8.2 2次多元回归模型的建立及显著性分析 采用Design Expert 8.0.6软件拟合得出OD的二次多元回归方程为OD=0.96+0.111+0.0372-0.153-2.5×10−412-0.05113-0.04623-0.4212-0.3622-0.2732。方差分析结果见表8,2=0.984 3,模型<0.000 1,失拟项=0.076 0>0.05。所以建立的模型具有极显著性水平,拟合度良好,未知因素干扰很小,可用于HD-PC-NPs处方工艺进行优化。另外,1、3、12、22、32是显著或极显著项。
2.8.3 响应面分析、优化与预测 固定TPGS与HD-PC用量比(1)、均质压力(2)及均质次数(3)3个因素之一,绘制另外2个因素对OD的三维曲面图,结果见图2。HD-PC-NPs的最佳处方为TPGS与HD-PC用量比为3.24∶1,均质压力95.29 MPa、均质次数为10.29次,预测最佳处方的粒径、PDI值和ζ电位绝对值分别为261.84 nm、0.117和−32.87 mV。

表7 Box-Behnken试验设计结果(n = 3)

表8 方差分析结果

图2 自变量与响应值的三维图
2.9 HD-PC-NPs处方验证
考虑到实际可操作性,调整TPGS与HD-PC用量比为3.2,均质压力95 mPa,均质次数为10次。平行制备3份HD-PC-NPs,分别测定粒径、PDI值和ζ电位,并计算偏差[偏差=(模型预测值-实际值)/模型预测值]。结果见表9,HD-PC-NPs粒径、PDI值和ζ电位的相对偏差均小于±5%,说明该数学模型具有良好的预测性和重现性。HD-PC-NPs的粒径及ζ电位图分别见图3。
2.10 HD-PC-NPs的TEM分析
取0.1 mL的HD-PC-NPs混悬液样品,用蒸馏水按照1∶40比例稀释,混匀后滴在铜网上,1.5%磷钨酸钠染色,自然晾干后置于TEM下观察HD-PC-NPs外貌(×15 000),结果见图4,所得HD-PC-NPs大体呈类球形或球形,粒子之间无粘连。
表9 实际值与预测值的比较(, n = 3)

图3 HD-PC-NPs的粒径分布(A)和ζ电位(B)图
2.11 HD-PC-NPs冻干粉研究
2.11.1 冻干保护剂种类及用量筛选 为增加HD-PC-NPs混悬液稳定性、便于储存及给药,故将其制备成冻干粉。取HD-PC-NPs混悬液,加入冻干保护剂,震荡混匀,于−65 ℃超低温冰箱中预冻24 h,迅速置于−25 ℃冷冻干燥机中冻干。以冻干粉外观、粒径及其PDI值进行评价,结果见表10,采用5%或7%甘露醇作为冻干保护剂时,冻干粉外观饱满,色泽均一,且粒径及PDI值变化相对较小。由于采用5%甘露醇时辅料用量相对较少,故选之作为HD-PC-NPs的冻干保护剂(外观见图5)。另测得HD-PC-NPs冻干粉中含橙皮苷的质量分数为(0.77±0.01)%。

图4 HD-PC-NPs的TEM图
表10 冻干保护剂种类及用量的考察(, n = 3)
2.11.2 冻干粉稳定性考察 取HD-PC-NPs冻干粉置于温度为40 ℃,湿度为65%恒温恒湿箱中,第6个月取适量冻干粉蒸馏水复溶后测得粒径为(284.53±24.09)nm,PDI值为0.243±0.029。HD-PC-NPs混悬液在相同条件下放置1个月即出现明显的沉淀现象,所以制备成冻干粉后可增加HD-PC-NPs的稳定性。

图5 HD-PC-NPs外观
2.12 HD-PC-NPs冻干粉的XRPD研究
取橙皮苷原料药、空白辅料(除不加橙皮苷外,其他辅料比例同HD-PC-NPs冻干粉)、物理混合物(橙皮苷与辅料比例同HD-PC-NPs冻干粉)和HD-PC-NPs冻干粉适量,按“2.5”项下条件分别进行XRPD扫描,结果见图6。物理混合物的XRPD图谱中仍可观察到橙皮苷原料药在8.0°、10.4°、10.5°、14.7°、14.9°、15.3°、15.8°等处的特征晶型峰。但在HD-PC-NPs冻干粉XRPD图谱中橙皮苷的特征晶型峰完全消失,仅可观察到辅料的XRPD图谱,证明橙皮苷存在状态发生了改变,转变为无定型物质。
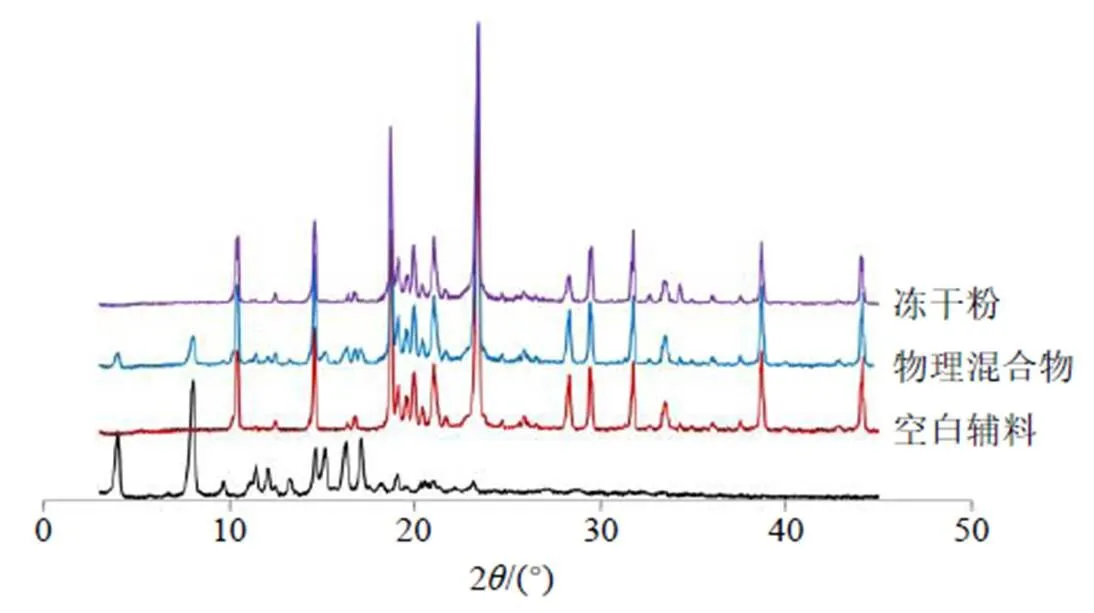
图6 HD-PC-NPs的XRPD图谱
2.13 溶解度考察
取过量的橙皮苷原料药、HD-PC、HD-PC-NPs冻干粉及其物理混合物(药物与辅料比例同HD-PC-NPs冻干粉)置于蒸馏水中,于25 ℃下震荡3 d(转速为100 r/min)。取上层混悬液,过0.45 μm微孔滤膜后测定,结果见表11。橙皮苷原料药溶解度仅为(6.32±0.12)μg/mL,HD-PC将其溶解度提高至2.48倍,而HD-PC-NPs将其溶解度提高至77.06倍。从物理混合物溶解度数据可以看出,辅料的增溶作用有限,提高幅度远低于HD-PC-NPs。
表11 溶解度测试结果(, n = 3)
2.14 HD-PC-NPs体外释药考察
橙皮苷溶解度受pH值影响较小[5]。测得橙皮苷在1.0% SDS水溶液中溶解度为(114.67±3.18)μg/mL,为达漏槽条件,故选择体积为900 mL,浓度为1.0% SDS水溶液作为释药介质[6]。取橙皮苷原料药、HD-PC、物理混合物(橙皮苷与辅料比例同HD-PC-NPs冻干粉)和HD-PC-NPs冻干粉样品(相当于橙皮苷含量20 mg),加入空白释药介质4 mL,并置于透析袋中(截留相对分子质量为8000~ 12 000),扎紧。设置温度为(37.0±0.5)℃,转速为75 r/min,于0、0.5、1.0、1.5、2.0、4.0、6.0、8.0、12.0、18.0、24.0、36.0 h取样4 mL,并补加空白介质4 mL。样品经8000 r/min离心(离心半径为8.6 cm)5 min后测定,结果见图7。橙皮苷原料药在36 h内累积释放率仅为8.12%,可能与橙皮苷原料药疏水性较强、颗粒较大有关。橙皮苷在物理混合物中的累积释放度略有提高(9.68%),可能与制剂中辅料增溶作用有关。HD-PC各点累积释放率得到明显改善,但36 h时仍未完全释药。而HD-PC-NPs冻干粉在4 h累积释放率达90%以上,显著提高了橙皮苷释药速率及释放度。
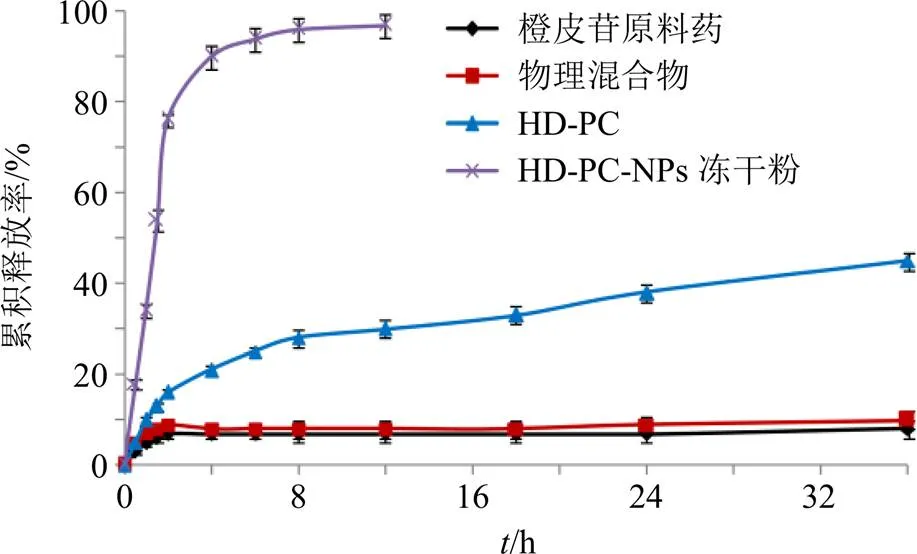
图7 体外药物释放曲线(, n = 3)
2.15 体内药动学研究
2.15.1 给药方案及样品采集 取禁食12 h的SD大鼠18只,随机分成橙皮苷原料药、HD-PC和HD-PC-NPs冻干粉3组,每组6只。采用0.5% CMC-Na水溶液配制灌胃液,剂量均为30 mg/kg(以橙皮苷含量计)。于0、0.25、0.50、1.00、1.50、2.00、3.00、4.00、6.00、8.00、10.00、12.00 h于眼眶后静脉取血约0.3 mL,至肝素化离心管中(肝素钠质量浓度为1.5 mg/mL),并于3500 r/min离心(离心半径为8.6 cm)3 min。移取上层血浆至空白离心管中,冷冻保存。
2.15.2 血浆样品处理[6,23]血浆样品解冻后精密移取100 μL,加入50 μL内标溶液和1 mL乙腈,涡旋3 min,于离心机中5000 r/min离心(离心半径为8.6 cm)5 min。取上层液至空白离心管中,40 ℃水浴下的氮气吹干。加入100 μL乙腈复溶,5000 r/min离心3 min,即得。
2.15.3 血浆对照品线性考察 精密取毛蕊异黄酮葡萄糖苷对照品,乙腈稀释至质量浓度为1200 ng/mL,作为内标溶液。乙腈稀释配制质量浓度为2000、1000、500、100、50、25 ng/mL的橙皮苷对照品溶液,各质量浓度取100 μL,分别加入内标溶液50 μL,混匀后于40 ℃水浴中氮气缓慢吹干。于残渣中分别加入空白血浆100 μL,后续按“2.14.2”项下处理,得血浆对照品溶液,进样测定,计算橙皮苷与内标峰面积比值。以橙皮苷质量浓度()对峰面积比值()进行线性回归,得橙皮苷回归方程=0.221 9+0.101 7,=0.994 2,所以线性范围为25~2000 ng/mL。
2.15.4 方法学考察 按“2.14.2”项下方法分别处理空白血浆和血浆样品,测定结果见图8,血浆内源性物质未干扰内标和橙皮苷色谱峰,专属性较高。取血浆样品于0、2、4、6、8、12 h进样,计算得橙皮苷和内标峰面积比值的RSD为9.18%,说明稳定性良好。取2000 ng/mL(高)、500 ng/mL(中)和25 ng/mL(低)血浆对照品溶液,连续测定6次,计算得橙皮苷和内标峰面积比值的RSD依次为6.97%、7.84%、9.37%,说明日内精密度良好。连续测定6 d,每天1次,计算得峰面积比值的RSD依次为4.49%、8.07%、6.14%,说明日间精密度良好。分别配制2000 ng/mL(高)、500 ng/mL(中)和25 ng/mL(低)的橙皮苷对照品溶液,按“2.14.2”项下方法处理后进样测定,并与配制质量浓度比较,计算回收率。结果显示,平均回收率为94.71%,RSD为5.90%,说明准确度较高。
2.15.5 药动学结果 橙皮苷原料药、HD-PC和HD-PC-NPs组的血药浓度-时间曲线,结果见图9。DAS2.0软件包拟合数据,结果见表12。橙皮苷制备成HD-PC后1/2延长至(4.71±0.66)h,max增加至(511.70±105.04)ng/mL,生物利用度提高至1.48倍。HD-PC-NPs组药动学行为发生了更大变化,主要药动学参数与橙皮苷原料药或物理混合物相比均具有极显著性差异(<0.01),其中max提前至(1.04±0.33)h,1/2延长至(5.69±0.82)h,max增加至(1 213.96±149.88)ng/mL,相对生物利用度提高至3.09倍,其提高幅度大于HD-PC,说明HD-PC-NPs促吸收作用更为显著。

图8 空白血浆(A)、血浆样品(B) 和橙皮苷+空白血浆(C)的HPLC图

图9 药-时曲线(, n = 6)
3 讨论
在制备PC时一般采用非质子溶剂作为反应介质,所以质子溶剂如甲醇、乙醇可能会干扰PC的形成,影响复合率[6]。四氢呋喃属于非质子溶剂,沸点较低,易于除去,且橙皮苷在四氢呋喃中溶解度相对较高,因而选为反应溶剂。PC中药物与磷脂摩尔比一般按照1∶1结合在一起[13-14],为使药物与磷脂完全形成PC,故在制备HD-PC时橙皮苷与磷脂的物质的量比控制为1∶1.2,此时的复合率基本达100%。
橙皮苷原料药难溶于水及各种有机溶剂。龙家英等[24]采用高速剪切联合高压均质法制备了橙皮苷纳米混悬剂,ζ电位为(−23.16±1.12)mV,绝对值不足30 mV,稳定性不高,可能与稳定剂种类过于单一有关[18]。前期研究发现,采用介质研磨法[25]直接将橙皮苷制备成NPs时,平均粒径达到(653.04±49.96)nm,粒径大小对药物体内吸收存在较大影响[21]。另外,橙皮苷热稳定性较差[26],采用介质研磨法制备HD-NPs时研磨介质之间的机械能转化成大量的热能,可能影响橙皮苷的稳定性。本研究将橙皮苷制备成HD-PC来提高其溶解度,为后续HD-PC-NPs的制备奠定基础。
本研究以HD-PC形式制得HD-PC-NPs粒径为(268.62±18.14)nm,为难溶性药物NPs处方工艺研究提供了新的思路。HD-PC-NPs处方中的磷脂发挥2方面的作用:①形成HD-PC后增加药物的溶解度,为制备HD-PC-NPs奠定基础;②磷脂是一种两亲性表面活性剂,可吸附于NPs表面提供负电荷,并与TPGS一同起到稳定作用[27]。

表12 主要药动学参数(, n = 6)
与橙皮苷比较:*<0.05**<0.01;与HD-PC比较:##<0.01
*< 0.05,**< 0.01hesperidin;##< 0.01HD-PC
HD-PC提高了橙皮苷溶解度及溶出度,但改善程度有限[6]。根据Ostwald-Freundlich方程,药物粒径对溶解度有很大影响,当粒径减小时,比表面积急剧增大,药物的溶解度会显著增大。同时也对药物溶出速率及累积溶出率产生积极影响。因而HD-PC-NPs与HD-PC相比,具有更高的溶解度、更快的溶出速率及更大的累积溶出度。
HD-PC将其生物利用度提高至1.48倍,但HD-PC-NPs的提高幅度更大(3.09倍)。HD-PC-NPs的max显著性提前,可能是由于HD-PC-NPs加快了橙皮苷的释药速率,缩短了经胃肠道黏膜吸收时间所致。HD-PC-NPs的max显著性增大可能是由于在HD-PC-NPs中引入PC技术后增加了药物的透膜能力所致,也与HD-PC-NPs极大提高了药物的累积溶出度有关。
HD-PC-NPs相对口服吸收生物利用度极显著性提高至3.09倍,可能是由于纳米药物比表面积急剧增大,有效增加了与胃肠道接触面;橙皮苷在HD-PC-NPs中以无定型形式存在,而无定型药物往往具有更高的生物利用度[28];纳米药物与胃肠道黏附性较强,有益于橙皮苷透膜吸收[29];橙皮苷在体内胃肠道稳定性较差,附着于HD-PC-NPs表面的稳定剂可能增加了药物的稳定性[19];HD-PC-NPs处方中的辅料如磷脂和TPGS等本身具有促进药物吸收的作用[12,30]。后续还需进一步完善HD-PC-NPs质量标准,评价HD-PC-NPs冻干粉的稳定性,继续对药效学和毒理学等进行研究,为橙皮苷纳米制剂研究开发提供更为全面、可靠的研究资料。
利益冲突 所有作者均声明不存在利益冲突
[1] Zhao J, Li Y L, Gao J F,. Hesperidin inhibits ovarian cancer cell viability through endoplasmic reticulum stress signaling pathways [J]., 2017, 14(5): 5569-5574.
[2] Shokoohi M, Khaki A, Shoorei H,. Hesperidin attenuated apoptotic-related genes in testicle of a male rat model of varicocoele [J]., 2020, 8(1): 249-258.
[3] 张恒, 饶坤林, 向韩. 橙皮苷药理活性研究进展 [J]. 中南药学, 2016, 14(10): 1097-1100.
[4] 刘沁如, 向茗, 瞿昊宇, 等. 陈皮黑茶颗粒提取工艺条件对降糖降脂活性成分的影响 [J]. 亚太传统医药, 2021, 17(12): 66-73.
[5] 林桂梅, 张国锋, 张建军. 枳实生品与麸炒品中4个黄酮类成分表观油水分配系数的比较研究 [J]. 中华中医药学刊, 2018, 36(6): 1417-1419.
[6] 牛晓磊, 贾润霞, 谈秀凤. 橙皮苷磷脂复合物固体分散体的制备、表征及其体内药动学研究 [J]. 中成药, 2020, 42(9): 2255-2259.
[7] 陈彦, 王晋艳, 贾晓斌, 等. 枳实中主要二氢黄酮类成分肠道吸收代谢及与药物相互作用的研究进展 [J]. 中草药, 2010, 41(9): 1564-1566.
[8] 高彩芳, 夏加璇, 朱颖, 等. 纳米技术在改善中药有效成分成药性中的应用 [J]. 中草药, 2018, 49(12): 2754-2762.
[9] 熊传爽, 田黎明, 洪怡, 等. 橙皮苷脂质体凝胶的制备及其透皮吸收研究 [J]. 中国医院药学杂志, 2022, 42(5): 511-518.
[10] 廖艳梅, 李小芳, 刘罗娜, 等. 橙皮苷纳米乳液的制备及其稳定性研究 [J]. 中草药, 2019, 50(10): 2312-2318.
[11] 凡小燕. 橙皮苷自微乳的制备与评价 [D]. 合肥: 安徽医科大学, 2011.
[12] 张留超, 刘勇华. 鞣花酸磷脂复合物的制备及其口服生物利用度研究 [J]. 中成药, 2021, 43(7): 1685-1690.
[13] 黄珊, 翟秉涛, 杨洁, 等. 根皮素磷脂复合物的制备、表征及体内外溶出行为评价 [J]. 中草药, 2021, 52(18): 5543-5551.
[14] 马记平, 刘丹花, 郝海军, 等. 根皮素磷脂复合物的制备及其体内药动学研究 [J]. 中成药, 2020, 42(6): 1577-1580.
[15] 祝上宾, 王英飒, 敬凡尘, 等. 熊果苷磷脂复合物的制备、表征及理化性质研究 [J]. 中草药, 2020, 51(22): 5698-5704.
[16] 邓向涛, 郝海军, 陈晓峰, 等. 木犀草素2种固体分散体制备、表征和大鼠体内药动学行为研究 [J]. 中草药, 2018, 49(24): 5787-5793.
[17] 郝海军, 张红芹, 贾幼智, 等. 采用制剂新技术提高中药磷脂复合物的溶出度和生物利用度研究进展 [J]. 中草药, 2013, 44(17): 2474-2479.
[18] 张亚林, 喻海洋, 黄涛. 大黄酸纳米混悬剂的制备及其体内药动学研究 [J]. 中成药, 2020, 42(11): 2829-2834.
[19] 毛艳婷, 陶兴茹, 张胜男, 等. 漆黄素纳米混悬剂和冻干粉末的制备及体外评价 [J]. 中国医院药学杂志, 2022, 42(4): 393-398.
[20] 黄甜甜, 沈一平, 路丽康, 等. 高载药量雷公藤红素纳米混悬剂的制备及体外评价 [J]. 中草药, 2020, 51(8): 2125-2133.
[21] 杭凌宇, 申宝德, 沈成英, 等. 不同粒径波棱甲素纳米混悬剂的制备及药动学研究 [J]. 中草药, 2021, 52(7): 1898-1905.
[22] 李怡静, 敖惠, 李好文, 等. 西瑞香素纳米混悬剂的制备及其体外抗肿瘤作用研究 [J]. 现代药物与临床, 2018, 33(2): 231-237.
[23] 相聪坤, 温俊霞, 闫宝环, 等. 陈皮汤中橙皮苷大鼠体内药动学分析方法的研究 [J]. 中国医院药学杂志, 2016, 36(18): 1542-1546.
[24] 龙家英, 李小芳, 王娴, 等. 茶皂素用于稳定橙皮苷纳米混悬剂及其机制研究 [J]. 药学学报, 2021, 56(11): 3159-3165.
[25] 申宝德, 连王权, 沈成英, 等. 微型化介质研磨法制备难溶性黄酮类化合物纳米混悬剂 [J]. 中草药, 2017, 48(21): 4413-4418.
[26] 文赤夫, 李国章, 董爱文, 等. 橙皮苷的提取及其稳定性分析 [J]. 生物质化学工程, 2006, 40(3): 37-40.
[27] 徐浩, 高艺璇, 王向涛. 槲皮素纳米混悬剂的制备、表征及抗乳腺癌研究 [J]. 中草药, 2019, 50(1): 42-51.
[28] 梁宇, 邢逞, 周启蒙, 等. 米非司酮3种晶型的制备及其在大鼠体内药动学研究 [J]. 中国新药杂志, 2020, 29(19): 2251-2259.
[29] 王小霞, 张智强. 鞣花酸纳米结构脂质载体处方优化和口服生物利用度研究 [J]. 中草药, 2021, 52(13): 3862-3871.
[30] 宋婷婷, 蔡荣珊, 王宏, 等. TPGS修饰的地榆皂苷I长循环脂质体的制备及质量评价 [J]. 中草药, 2021, 52(12): 3522-3529.
Preparation, characterization andoral pharmacokinetics of hesperidin phospholipids complex nanosuspensions
LI Xi1, 2, 3, ZHANG Wen-zhou1, 2, 3, HAO Hai-jun4
1. Department of Pharmacy, Affiliated Cancer Hospital of Zhengzhou University and Henan Cancer Hospital, Zhengzhou 450000, China 2. Henan Engineering Research Center for Tumor Precision Medicine and Comprehensive Evaluation, Zhengzhou 450000, China 3. Henan Provincial Key Laboratory of Anticancer Drug Research, Zhengzhou 450000, China 4. Shanghai Institute of Traditional Chinese Medicine, Shanghai 201401, China
To prepare hesperidin phospholipids complex (HD-PC) nanosuspensions (HD-PC-NPs) and study its pharmacokinetics behavior in SD rats.Hesperidin was prepared into HD-PC to enhance its solubility. HD-PC-NPs were prepared by nano-precipitation and high pressure homogenization method. Based on single factor experiments, amounts ratio of stabilizer to HD-PC, homogenization pressure and frequency were used as influencing factors, overall values (OV) of average particles size, polydispersion index (PDI) and ζ potential were employed as evaluation indexes, the formulation of HD-PC-NPs was optimized by Box-Behnken design-response surface method. Lyophilized powder of HD-PC-NPs was prepared. Morphology of HD-PC-NPs was observed by transmission electron microscope (TEM). Dialysis bag method was employed to investigate the drug release. SD rats were divided into hesperidin suspension group, HD-PC group and HD-PC-NPs group. Hesperidin concentration in plasma was analyzed by HPLC, main pharmacokinetic parameters and relative oral bioavailability were also calculated.Optimal preparation of HD-PC-NPs was as follows: amounts ratio of stabilizer to HD-PC was 3.2, homogenization pressure was 95 MPa, homogenization frequencies was 10 times and temperature was 50 ℃. Lyophilized powder has a full appearance prepared by 5% mannitol. The morphology of HD-PC-NPs was spherical or quasi-spherical. Average particle size, PDI and ζ potential of HD-PC-NPs were (268.62 ± 18.14) nm, 0.122 ± 0.013 and (−31.79 ± 1.37) mV, respectively. Solubility of hesperidin was enhanced to 77.06 times and cumulative dissolution in 6 h was increased to 94.68% by HD-PC-NPs. Themaxof HD-PC-NPs was advanced significantly,1/2was prolonged to (5.69 ± 0.82) h,maxwas increased to (1 213.96 ± 149.88) ng/mL and the oral bioavailability was enhanced to 3.09 times.HD-PC-NPs could enhance solubility of hesperidin, and promote dissolutionand absorption.
hesperidin; phospholipids complex; nanosuspensions; Box-Behnken design-response surface method; bioavailability; nano-precipitation and high pressure homogenization method
R283.6
A
0253 - 2670(2022)24 - 7740 - 11
10.7501/j.issn.0253-2670.2022.24.012
2022-07-24
河南省二〇二一年科技发展计划(2002310325);上海市科委项目(21S21903400)
李 茜(1989—),女,硕士,主管药师,主要从事药动学、医院药学研究。Tel: (0371)65588378 E-mail: hnzlyylixi@126.com
张文周(1969—),男,学士,主任药师,主要从事医院药学研究。Tel: (0371)65588378 E-mail: hnzzzwz@sina.com
[责任编辑 郑礼胜]
