光甘草定通过调节ERK/IRS-1和PI3K/Akt信号通路改善HepG2细胞的胰岛素抵抗
2022-12-28李德锋樊金玲任国艳
李德锋,樊金玲,杜 琳,任国艳
• 药理与临床 •
光甘草定通过调节ERK/IRS-1和PI3K/Akt信号通路改善HepG2细胞的胰岛素抵抗
李德锋,樊金玲*,杜 琳,任国艳
河南科技大学食品与生物工程学院,河南 洛阳 471023
探索光甘草定改善肝细胞胰岛素抵抗(insulin resistance,IR)的作用和机制。通过高胰岛素诱导人肝癌HepG2细胞建立IR模型,采用葡萄糖氧化酶法检测细胞的葡萄糖消耗量及生成;荧光标记法检测葡萄糖摄取量;蒽酮法检测糖原含量;ELISA检测葡萄糖代谢关键酶的活性;Western blotting检测磷脂酰肌醇3-激酶/蛋白激酶B(phosphatidylinositol 3-kinase/protein kinase B,PI3K/Akt)、细胞外调节蛋白激酶/胰岛素受体底物-1(extracellular regulated protein kinase/insulin receptor substrate-1,ERK/IRS-1)信号通路相关蛋白以及葡萄糖转运蛋白4(glucose transporter 4,GLUT4)的表达。采用分子对接技术研究光甘草定和ERK分子间的相互作用。光甘草定显著增加IR-HepG2细胞的葡萄糖消耗和摄取(<0.05);通过显著提高糖原合成酶(glycogen synthase,GS)、葡萄糖激酶(glucokinase,GCK)和丙酮酸激酶(pyruvate kinase,PK)活性(<0.05、0.01),促进IR-HepG2细胞的糖原合成和糖酵解;通过显著减弱磷酸烯醇丙酮酸羧激酶(phosphoenolpyruvate carboxykinase,PEPCK)和葡萄糖-6-磷酸酶(glucose-6-phosphatase,G6Pase)的活性(<0.05),抑制IR-HepG2细胞的糖异生。IR-HepG2细胞经光甘草定处理后,Akt、糖原合成酶激酶-3β(glycogen synthase kinase-3β,GSK-3β)和叉头框蛋白O1(forkhead boxing protein O1,FOXO1)的磷酸化水平得到显著恢复(<0.01),而这种作用被PI3K的抑制剂LY294002所逆转(<0.01)。同时,光甘草定显著促进GLUT4向质膜的易位(<0.01)。光甘草定显著降低IR-HepG2细胞的ERK和IRS的磷酸化水平(<0.01),还可作为ERK的I1/2型抑制剂。光甘草定通过抑制ERK/IRS-1通路,激活PI3K/Akt信号通路,修复IR-HepG2细胞的糖代谢紊乱,缓解IR症状。
胰岛素抵抗;ERK/IRS-1;PI3K/Akt;光甘草定;HepG2细胞;糖代谢;葡萄糖摄取
2型糖尿病(type 2 diabetes mellitus,T2DM)是一种慢性疾病,以糖脂代谢紊乱为特征[1]。胰岛素抵抗(insulin resistance,IR)是导致T2DM的主要诱因。IR发生时,肝脏相对其他靶器官更早呈现IR症状[2],是受IR影响最严重的组织之一,表现为胰岛素信号转导受损[主要是磷脂酰肌醇-3-激酶/蛋白激酶B(phosphatidylinositol-3-kinase/protein kinase B,PI3K/Akt)通路异常,PI3K/Akt途径是介导胰岛素刺激细胞摄取利用葡萄糖的主要途径[3]],一方面导致肝脏的葡萄糖摄取减少,另一方面导致糖酵解和糖原合成能力减弱、肝糖原的分解和糖异生加强,使得肝糖原输出增加;最终造成肝脏调节血糖的能力减弱,血糖代谢出现紊乱。
光甘草定是光果甘草L中特有的疏水性异黄烷,具有抗氧化、抗炎、抗肿瘤、抗动脉粥样硬化、神经保护等多种生物活性[4]。研究发现,光甘草定对动物模型表现出显著的降血糖和抗糖尿病活性[5-6],能够促进正常大鼠L6骨骼肌细胞对葡萄糖的摄取,预防骨骼肌细胞的葡萄糖不耐受[7]。研究者先后采用MC3T3-E1成骨细胞[8]、J774A.1小鼠巨噬细胞[9]、THP-1人类白血病单核细胞[10]、3T3-L1前脂肪细胞[11]等探索了光甘草定抗糖尿病的作用机制,目前还未发现关于光甘草定对肝细胞糖代谢的影响及机制的报道。
细胞外调节蛋白激酶(extracellular regulated protein kinase,ERK)是丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)家族最主要的成员,主要调节细胞凋亡、增殖和分化[12]。研究表明ERK信号通路与胰岛素信号级联和T2DM的发展密切相关[13-14]。过度激活的ERK可以诱导胰岛素受体底物-1(insulin receptor substrate-1,IRS-1)在Ser307/612/632/636处发生丝氨酸磷酸化[15-16],抑制其酪氨酸磷酸化,降低IRS-1的活性,阻断胰岛素信号的转导,引起IR的发生[17]。ERK信号通路是胰岛素信号转导的途径之一。正常生理条件下,胰岛素刺激可激活PI3K/Akt通路和ERK通路[18],前者调节葡萄糖代谢,包括葡萄糖摄取和糖生成[19];后者主要调节细胞的增殖和分化[20],同时对胰岛素激活PI3K/Akt途径起到负反馈调节的作用。ERK途径还受胰岛素以外的信号激活,如生长因子、细胞因子、病毒、G-蛋白偶联受体配体、致癌物等[12],这些刺激物在病理条件下会使ERK过度激活,从而损害胰岛素信号的转导。因此,PI3K/Akt和ERK这2种信号途径的平衡被认为是胰岛素敏感性的关键调控器[14,21]。本课题组前期采用网络药理学研究了光甘草定改善T2DM潜在的作用机制,通过对“疾病-靶点-通路”网络的拓扑分析筛选出ERK2是光甘草定潜在的作用靶点之一。
本研究通过高胰岛素诱导人肝癌HepG2细胞建立IR模型,通过测定光甘草定对HepG2细胞胰岛素抵抗(IR-HepG2)模型葡萄糖消耗和摄取、糖酵解、糖原合成、糖异生以及糖代谢途径中关键酶的活性,探讨光甘草定对肝细胞葡萄糖代谢紊乱的修复作用;通过测定光甘草定对ERK/IRS-1和PI3K/Akt通路的关键信号节点和下游底物蛋白的表达及磷酸化水平,探讨光甘草定修复肝细胞葡萄糖代谢紊乱的调控途径;并采用分子对接技术研究光甘草定与ERK的分子间相互作用,旨在从分子水平揭示ERK作为光甘草定作用靶点的可能性。为进一步推动光甘草定作为功能性降糖活性成分在功能食品、膳食补充剂和药品中的应用提供实验依据。
1 材料
1.1 细胞
HepG2细胞由陕西师范大学赠送。
1.2 药品与试剂
光甘草定(质量分数为90%,批号GF2020071501)购自洛阳蓝斯利科技有限公司;DMEM高糖培养基(25 mmol/L,批号AG29714104)、DMEM低糖培养基(5.5 mmol/L,批号AG29694193)购自美国Hyclone公司;DMEM无糖培养基(批号PM150270)购自武汉普诺赛生命科技有限公司;南美胎牛血清(fetal bovine serum,FBS,批号CCS30009)购自上海迪奥生物有限公司;MTT(批号298-93-1)购自上海蓝季生物公司;胰岛素(批号11061-68-0)购自美国Sigma公司;盐酸二甲双胍(批号1115-7-4)购自上海施贵宝制药有限公司;荧光-葡萄糖类似物2-NBDG(批号M6327)购自美国Abmole公司;葡萄糖检测试剂盒(批号A154-1-1)、磷酸烯醇丙酮酸羧激酶(phosphoenolpyruvate carboxykinase,PEPCK)测定试剂盒(批号A131-1-1)、丙酮酸激酶(pyruvate kinase,PK)测定试剂盒(批号A076-1-1)购自南京建成生物工程研究所有限公司;糖原合成酶(glycogen synthase,GS)ELISA试剂盒(批号YB-GS-Hu)、葡萄糖激酶(glucokinase,GCK)ELISA试剂盒(批号YB-GCK-Hu)、葡萄糖-6-磷酸酶(glucose-6-phosphatase,G6Pase)ELISA试剂盒(批号YB-G6Pase-Hu)购自上海钰博生物技术有限公司;糖原测定试剂盒(批号BC0345)购自北京索乐博科技有限公司;BCA蛋白定量试剂盒(批号E-BC-K318-M)购自武汉精英生物科技有限公司;细胞膜和细胞质蛋白提取试剂盒(批号P0033)购自上海碧云天生物技术有限公司;IRS-1抗体(批号AP70586)、Akt抗体(批号AP7028B)、磷酸化Akt(Ser473,phosphorylated Akt,p-Akt)抗体(批号AP3434a)、糖原合成酶激酶-3β(glycogen synthase kinase-3β,GSK-3β)抗体(批号AP60301)、磷酸化GSK-3β(Ser9,phosphorylated GSK-3β,p-GSK-3β)抗体(批号AP67217)、叉头框蛋白O1(forkhead boxing protein O1,FOXO1)抗体(批号AP60814)、磷酸化FOXO1(Ser256,phosphorylated FOXO1,p-FOXO1)抗体(批号Ap67045)、葡萄糖转运蛋白4(glucose transporter 4,GLUT4)抗体(批号AP60050)、ERK抗体(批号AM2189b)购自美国Abcepta公司;磷酸化IRS-1(Ser307,phosphorylated IRS-1,p-IRS-1)抗体(批号Ab5599)购自英国Abcam公司;磷酸化ERK(Thr202/Tyr204,phosphorylated ERK,p-ERK)抗体(批号AF1015)购自美国Affinity公司;β-actin抗体(批号66009-1-lg)购自美国Proteintech公司;Na+, K+-ATPase抗体(批号ABL1141)购自美国Abbine公司;ERK途径抑制剂PD98059(批号HY-12028)、PI3K抑制剂LY294002(批号HY-10108)购自Med Chem Express公司;山羊抗小鼠二抗(批号CW0102S)、山羊抗兔二抗(批号CW0103S)购自康为世纪生物科技股份有限公司。
1.3 仪器
SW-CJ-2FD型双人单面净化工作台(苏州净化设备有限公司);CO2培养箱(金西盟人工智能有限责任公司);UV-4800型紫外可见分光光度计(上海尤尼柯仪器有限公司);倒置荧光显微镜(日本Olympus公司);多功能酶标仪(美国Bio-Rad公司);DYY-6C型稳压稳流电泳仪(北京六一仪器厂);5200型Multi全自动化学发光成像分析系统(上海天能科技有限公司)。
2 方法
2.1 细胞培养和IR-HepG2模型的建立
HepG2细胞用含10% FBS和1%双抗的DMEM低糖培养基,于37 ℃、5% CO2细胞培养箱中培养;用0.25%胰蛋白酶进行消化、传代。取对数生长期的细胞以5×104个/mL接种于96孔板中,每孔200 μL,继续培养24 h;用无血清的DMEM高糖培养基替换DMEM低糖培养基再培养12 h;弃去旧培养基,用PBS冲洗细胞3次,然后用含有5×10−6mol/L胰岛素的DMEM高糖培养基(含10% FBS)培养36 h;检测各孔细胞培养基上清液中葡萄糖含量,计算葡萄糖消耗量。造模细胞的葡萄糖消耗量显著低于正常细胞,则表明IR模型建立成功。
2.2 IR-HepG2模型的稳定时间
模型建立成功后,将正常细胞组和模型组同时置于不含胰岛素的DMEM高糖培养基中继续培养24、36、48 h;检测各孔细胞培养基上清液中葡萄糖含量,计算葡萄糖的消耗量。模型组细胞的葡萄糖消耗量显著低于正常细胞,则表明IR模型在相应的时间内保持稳定。
2.3 细胞活力的测定
取HepG2细胞和建模成功的IR-HepG2细胞,用PBS清洗1~2次,然后每孔加入200 μL由DMEM高糖培养基(含10% FBS)配成的不同质量浓度的光甘草定(0、0.1、0.5、1.0、5.0、10.0、15.0 μg/mL),于37 ℃、5% CO2细胞培养箱中孵育24 h后,用PBS冲洗细胞1~2次,采用MTT法测定细胞活性。
2.4 糖代谢相关实验
2.4.1 分组及处理 设置对照组、模型组及光甘草定低、中、高剂量(0.1、1.0、10.0 μg/mL)组和二甲双胍(0.5 mmol/L)组。对照组细胞用不含胰岛素的DMEM高糖培养基培养;模型组、光甘草定各剂量组和二甲双胍组均按“2.1”项下方法诱导IR。细胞用PBS冲洗3次后,对照组和模型组加入DMEM高糖培养基,光甘草定各剂量组加入DMEM高糖培养基配制的不同质量浓度的光甘草定溶液;二甲双胍(0.5 mmol/L)组加入由DMEM高糖培养基配制的二甲双胍溶液。各组均在37 ℃、5% CO2细胞培养箱中继续培养24 h。
2.4.2 葡萄糖消耗测定 96孔板中(HepG2细胞5×104个/mL)进行“2.4.1”项下实验分组及处理,并设置空白孔。处理结束后,收集每孔的培养液,用葡萄糖检测试剂盒(葡萄糖氧化酶法)检测培养液中的葡萄糖含量,计算各孔的葡萄糖消耗量。
葡萄糖消耗量=空白孔葡萄糖含量-每孔葡萄糖含量
2.4.3 葡萄糖摄取测定 24孔板中(HepG2细胞1×105个/mL)进行“2.4.1”项下实验分组及处理,其中光甘草定处理组只选取高剂量组。处理结束后,弃去培养基,用PBS洗板2次;然后加入50 μmol/L的2-NBDG溶液,并在37 ℃、5% CO2细胞培养箱中孵育30 min;弃去2-NBDG溶液,用冷PBS清洗2~3次;在倒置荧光显微镜下观察,获得荧光图像,并利用Image J软件对荧光强度进行定量分析。
2.4.4 糖原含量测定 6孔板中(HepG2细胞5×105个/mL)进行“2.4.1”项下实验分组及处理,其中光甘草定处理组只选取高剂量组。处理结束后,收集细胞,然后按照糖原测定试剂盒(蒽酮法)中的操作方法测定各组细胞中的糖原含量。
2.4.5 葡萄糖生成测定 6孔板中进行“2.4.1”项下实验分组及处理,其中光甘草定处理组只选取高剂量组。处理结束后,弃去培养基,用PBS洗板2次;加入含2 mmol/L丙酮酸钠和20 mmol/L乳酸钠的无糖DMEM培养基,培养4 h。然后收集各孔的培养基和细胞,按照葡萄糖测定试剂盒(葡萄糖氧化酶法)中的步骤检测各组葡萄糖含量,并将葡萄糖含量标准化为细胞蛋白浓度。
2.4.6 糖代谢关键酶活性测定 6孔板中进行“2.4.1”项下实验分组及处理,其中光甘草定处理组只选取高剂量组。处理结束后,弃去培养基,用PBS洗板2次,收集细胞。按照ELISA试剂盒说明书分别测定GCK、GS和G6Pase的活性,按照PK和PEPCK检测试剂盒说明书分别测定PK和PEPCK的活性。
2.5 PI3K/Akt和ERK/IRS-1途径中蛋白表达及磷酸化的检测
2.5.1 分组及处理 在6孔板中(HepG2细胞1×106个/mL)进行分组及处理。设置对照组、模型组、光甘草定高剂量组、高剂量光甘草定+LY294002组和PD98059抑制剂组。对照组用不含胰岛素的DMEM高糖培养基培养;模型组、光甘草定高剂量组、高剂量光甘草定+LY294002组、PD98059组均按“2.1”项下方法诱导IR。细胞用PBS冲洗3次后,对照组、模型组、光甘草定高剂量组细胞的处理方法同“2.4.1”项;高剂量光甘草定+LY294002组和PD98059组先用DMEM高糖培养基配成的LY294002(10 μmol/L)或PD98059(10 μmol/L)溶液预处理细胞1 h,然后加入DMEM高糖培养基配成的光甘草定+LY294002样品液或PD98059溶液处理24 h。处理结束后,弃去培养基;各组均加入含100 nmol/L胰岛素的DMEM高糖培养基,并在培养箱中刺激20 min;弃去培养基,然后用冷PBS洗板1~2次,收集细胞。
2.5.2 PI3K/Akt途径中蛋白表达及其磷酸化的检测 对照组、模型组、光甘草定高剂量组和高剂量光甘草定+LY294002组收集细胞后,通过Western blotting法检测PI3K/Akt信号通路中Akt、GSK-3β和FOXO1蛋白的表达及其磷酸化。
2.5.3 GLUT4易位的检测 对照组、模型组、光甘草定高剂量组收集细胞后,根据细胞膜和细胞质蛋白提取试剂盒中的步骤提取细胞膜蛋白和细胞质蛋白,然后通过Western blotting法分别检测细胞膜和细胞质中GLUT4的表达。
2.5.4 ERK/IRS-1途径中蛋白表达及其磷酸化的检测 对照组、模型组、光甘草定高剂量组和PD98059抑制剂组收集细胞后,通过Western blotting法检测ERK和IRS-1蛋白表达及其磷酸化。
2.5.5 Western blotting分析 收集细胞后,用RIPA缓冲液裂解细胞,离心后取上清液,用BCA蛋白定量试剂盒测量蛋白浓度。蛋白样品经十二烷基硫酸钠-聚丙烯酰胺凝胶电泳,转至PVDF膜,用脱脂牛奶封闭1.5 h,加入一抗,4 ℃孵育过夜;用TBST缓冲液洗涤后,加入二抗,37 ℃孵育2 h,使用全自动化学发光成像分析系统进行化学发光成像,使用成像仪自带图像分析软件GIS 1D对曝光结果进行分析。
2.6 分子对接
分别从PDB蛋白质结构数据库和PubChem数据库中下载受体的晶体结构和配体的化学结构,ERK2对应的PDB ID为4ZZN,采用AutoDock Tools工具对上述蛋白受体和配体进行常规处理,再用其Autogrid模块得到对接活性位点,运行程序进行分子对接,并利用Pymol软件绘制光甘草定与ERK2分子对接图。
2.7 数据统计分析
3 结果
3.1 光甘草定对IR-HepG2细胞葡萄糖消耗的影响
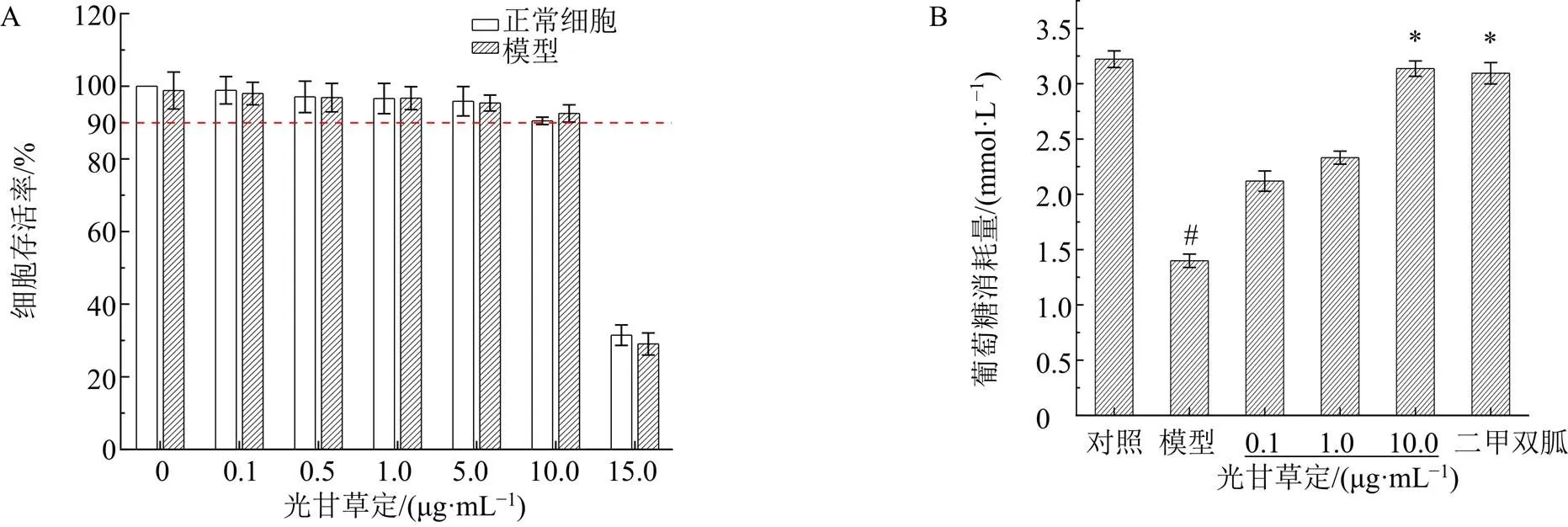
葡萄糖的消耗是细胞摄取葡萄糖和外排葡萄糖的净结果,直接反映细胞对外源葡萄糖的利用情况。首先在IR-HepG2细胞模型建立并可在48 h内保持稳定的基础上,分析光甘草定的细胞毒作用,测定了光甘草定对IR-HepG2细胞葡萄糖消耗量的影响,如图1-A所示,当光甘草定质量浓度为0.1~10.0 μg/mL时,HepG2细胞和IR-HepG2细胞的存活率均在90%以上,表明光甘草定在此质量浓度范围内对HepG2细胞和IR-HepG2细胞没有细胞毒性。选取0.1、1.0和10.0 μg/mL光甘草定分别孵育IR-HepG2细胞24 h,如图1-B所示,3个剂量的光甘草定均不同程度地增加IR-HepG2细胞的葡萄糖消耗,且呈剂量相关性,其中,光甘草定高剂量组的细胞葡萄糖消耗量显著高于模型组(<0.05),与二甲双胍阳性对照组(0.5 mmol/L)无显著差异,表明10 μg/mL的光甘草定可有效改善IR-HepG2细胞对外源葡萄糖的利用。后续实验的光甘草定质量浓度均选取10 μg/mL。
3.2 光甘草定对IR-HepG2细胞葡萄糖摄取的影响
采用2-NBDG荧光标记物检测光甘草定对IR-HepG2细胞摄取葡萄糖的影响,如图2所示,与对照组比较,模型组2-NBDG摄取量显著降低(<0.01);光甘草定处理显著提高了IR-HepG2细胞的葡萄糖摄取量(<0.05),效果与二甲双胍阳性对照组(0.5 mmol/L)无显著差异。
与对照组比较:#P<0.05 ##P<0.01 ###P<0.001;与模型组比较:*P<0.05 **P<0.01,下图同

图2 光甘草定对IR-HepG2细胞葡萄糖摄取的影响(, n = 3)
3.3 光甘草定对IR-HepG2细胞葡萄糖代谢的影响
通过测定光甘草定对IR-HepG2细胞的糖原合成量、葡萄糖生成量以及关键酶活性的影响,研究光甘草定对发生IR的肝细胞中葡萄糖代谢的调节作用。
3.3.1 光甘草定促进IR-HepG2细胞的糖酵解 PK和GCK是糖酵解途径中的2个关键限速酶。如图3所示,模型组PK和GCK的活性与对照组相比均显著降低(<0.01);而光甘草定处理显著提高了IR细胞的PK和GCK活性(<0.05、0.01),提示光甘草定可以通过上调IR-HepG2细胞中PK和GCK的活性促进糖酵解。
3.3.2 光甘草定提高IR-HepG2细胞的糖原合成 IR发生时,肝细胞的肝糖原合成受阻、而糖原分解加快,是造成肝脏葡萄糖生成量增加的一个重要原因。GS是糖原合成的关键酶。如图4所示,模型组的GS活性和糖原含量均显著低于对照组(<0.01);经光甘草定处理后,GS活性和糖原含量均显著提高(<0.05),表明光甘草定可以通过上调IR-HepG2细胞中GS的活性促进糖原合成。
3.3.3 光甘草定抑制IR-HepG2细胞的糖异生 糖异生是T2DM患者空腹肝葡萄糖生成增加的主要原因[22]。PEPCK、G6Pase是糖异生途径中的关键限速酶。如图5所示,模型组PEPCK、G6Pase的活性和葡萄糖的生成量均显著高于对照组(<0.05、0.01);而在光甘草定作用下,这种增加被显著抑制(<0.05)。表明光甘草定可以通过下调IR-HepG2细胞的G6Pase和PEPCK的活性来抑制糖异生。
3.4 光甘草定通过激活PI3K/Akt信号通路调节IR-HepG2细胞的葡萄糖代谢
胰岛素信号转导受损,即PI3K/Akt通路异常是肝脏发生IR导致葡萄糖代谢失衡的主要原因。通过测定光甘草定对该通路的关键信号节点Akt以及Akt的下游底物GSK-3β和FOXO1的表达及磷酸化水平,探讨光甘草定对PI3K/Akt通路的修复作用以及调节糖原合成、葡萄糖生成的机制。如图6所示,与对照组比较,模型组Akt、GSK-3β和FOXO1的磷酸化被显著抑制(<0.001),光甘草定处理则显著提高了三者的磷酸化水平(<0.01),但这种作用被PI3K抑制剂LY294002显著逆转(<0.01)。表明光甘草定对IR-HepG2细胞葡萄糖代谢紊乱的修复作用是由PI3K介导的,通过激活肝细胞中PI3K/Akt信号通路,一方面促进了GSK-3β的磷酸化从而促进了糖原合成,另一方面增强了FOXO1的磷酸化从而抑制了糖异生。

图3 光甘草定对IR-HepG2细胞PK和GCK活性的影响(, n = 3)

图4 光甘草定对IR-HepG2细胞糖原合成和GS活性的影响(, n = 3)

图5 光甘草定对IR-HepG2细胞糖异生的影响(, n = 3)

与光甘草定组比较:&&P<0.01
3.5 光甘草定促进IR-HepG2细胞GLUT4的质膜易位
胰岛素通过增加GLUT4从细胞内的储存囊泡到质膜的易位来促进葡萄糖摄取,是降低血糖水平的主要机制之一[23]。为了确定光甘草定对GLUT4易位的影响,对光甘草定处理后的IR-HepG2细胞进行质、膜组分的Western blotting检测,如图7所示,β-actin和Na+, K+-ATPase分别在质膜和细胞质中几乎检测不到,表明质膜和细胞质成分分离较好。与对照组比较,模型组细胞质膜上的GLUT4表达显著降低(<0.001),而细胞质中GLUT4的表达显著升高(<0.001);光甘草定处理则显著提高了GLUT4在IR-HepG2细胞质膜中的表达(<0.01),显著降低其在细胞质中的表达(<0.01)。表明光甘草定可以显著促进细胞内储存GLUT4的囊泡向质膜的易位,从而刺激IR-HepG2细胞的葡萄糖摄取。
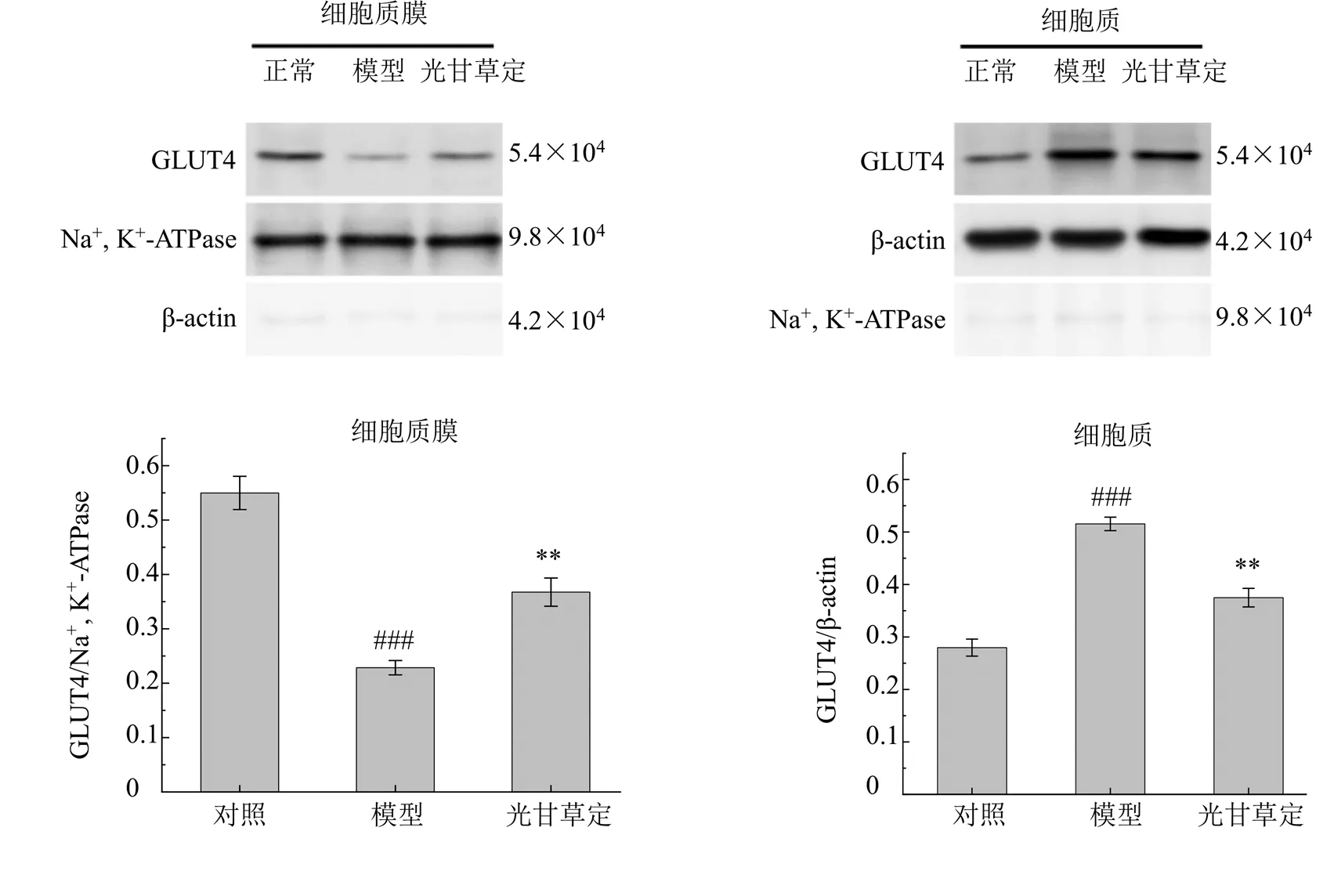
图7 光甘草定对IR-HepG2细胞中GLUT4易位的影响(, n = 3)
3.6 光甘草定对IR-HepG2细胞ERK/IRS-1信号通路的影响
过度激活的ERK可以诱导IRS-1在Ser307/612/ 632/636处发生丝氨酸磷酸化[15-16],阻断胰岛素信号的转导。通过测定光甘草定对ERK和IRS-1的表达及磷酸化水平,进一步探讨了光甘草定修复PI3K/Akt通路的途径,如图8所示,与对照组比较,模型组ERK和IRS-1的磷酸化水平均显著升高(<0.001);PD98059处理有效阻断了ERK的磷酸化(<0.01),同时显著抑制了IRS-1的磷酸化(<0.01);与PD98059处理相类似,光甘草定处理同样显著降低了IR-HepG2细胞中ERK和IRS-1的磷酸化(<0.01)。表明在肝细胞发生IR时,光甘草定可以通过抑制ERK/IRS-1通路的激活,修复PI3K/Akt途径的损伤,促进胰岛素信号的转导。
3.7 光甘草定与ERK2的分子对接结果
本课题组前期的网络药理学研究结果表明ERK2是光甘草定潜在的作用靶点之一。采用分子对接技术研究光甘草定与ERK之间的分子相互作用,旨在从分子水平揭示光甘草定作为ERK抑制剂的可能性。
ERK2具有由端和端卷曲形成的双叶结构。端主要由5个β折叠(β1~β5)、1个αC螺旋和1个甘氨酸富集环结构组成,它们对ATP的定位具有重要作用。其中,β1和β2-折叠占据了部分腺嘌呤结合口袋;β3折叠通常包含1个保守的AXK序列,其赖氨酸(Lys)将ATP的α-和β-磷酸盐耦合到αC-螺旋上;甘氨酸富集环位于β1、β2-折叠之间,是端中最灵活的部分,有助于定位ATP的β-和γ-磷酸盐以进行催化作用。β3-Lys和αC-谷氨酸(Glu)之间形成的1个盐桥是激酶处于活性状态的先决条件,称为αCin构象;如果这种盐桥缺失,则表明该激酶是非活性的,称为αCout构象。端主要由6个α螺旋和4股较短的β折叠(β6~β9)组成,在β6~β9折叠中包含了与ATP向ERK1/2底物磷酸化转移有关的大部分催化残基。端和端通过一段柔性的铰链区连接,该铰链区域与ATP结合位点部分重叠,也被称为“活化loop”。“活化loop”一般由20~30个氨基酸残基组成,从保守的DFG基序(Asp-Phe-Gly)开始,以保守的APE序列(Ala-Pro-Glu)结束。在活性蛋白激酶中,DFG-天冬氨酸(Asp)侧链(ERK2 Asp165)靠近ATP结合位点,称为DFG-in构象;在非活性蛋白激酶中,DFG-Asp侧链远离ATP结合位点,称为DFG-out构象[24]。DFG-in/αCin表示活性蛋白激酶;DFG-in/αCout、DFG-out/αCin和DFG-out/αCout表示非活性蛋白激酶[25]。依据蛋白激酶的构象,将其抑制剂分为I、II、I1/2和III型[26-27]。I型抑制剂与活性蛋白激酶的DFG-in构象结合,可逆的结合到ATP结合位点;II型抑制剂与非活性的蛋白激酶的DFG-out构象结合;I1/2型抑制剂与非活性蛋白激酶的DFG-in构象结合(与II型抑制剂的DFG-out构象相反),占据了ATP部分结合位点;III型抑制剂与蛋白激酶的变构位点结合,不影响ATP的结合。Liao[28]和van Linden等[29]将蛋白激酶端和端之间的区域划分为“Front cleft”“Gate area”和“Back cleft”。“Front cleft”包括铰链区部分残基、腺嘌呤结合口袋和甘氨酸富集环;“Gate area”包括端叶的β3折叠和“活化loop”的近端部分。
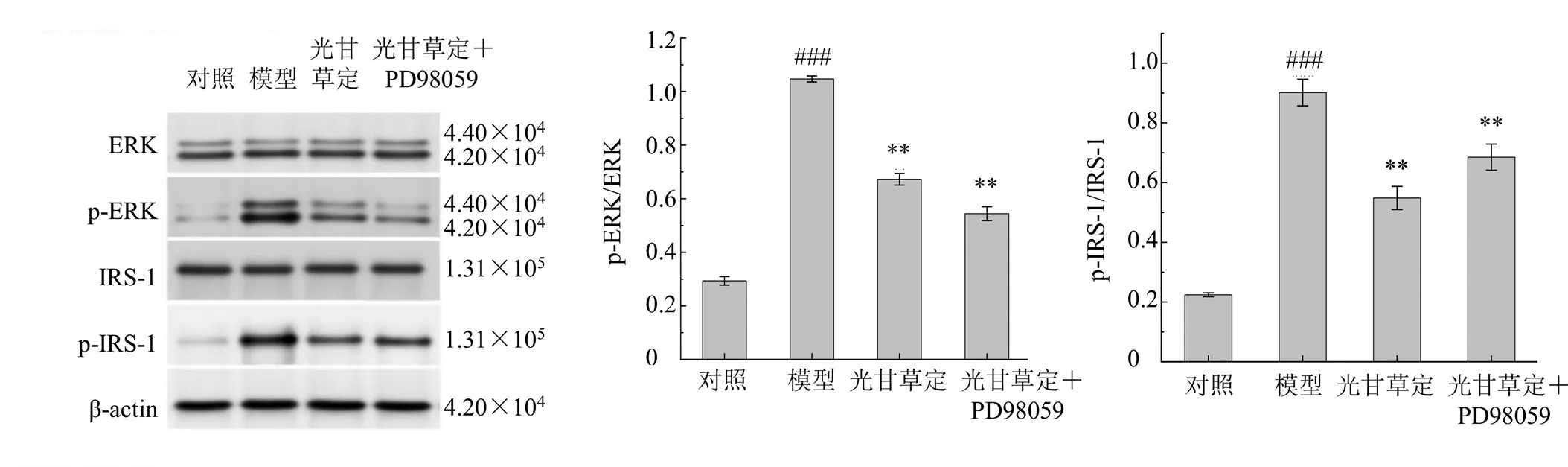
图8 光甘草定对IR-HepG2细胞中ERK/IRS-1通路相关蛋白表达的影响(, n = 3)
光甘草定与ERK2的结合能为−27.78 kJ/mol,说明光甘草定与ERK2能自发且稳定地结合,具有较好的亲和力。光甘草定结合在了ERK2的“Front cleft”和“Gate area”处。从图9-A中可以观察到DFG-Asp165靠近ATP结合位点;β3-Lys52和αC-Glu69之间没有形成盐桥,这表明与光甘草定结合的ERK2具有DFG-in/αCout构象。因此,该激酶是非活性状态。光甘草定的羟基分别与甘氨酸富集环上Ala33、αC螺旋上Tyr62形成了氢键;与甘氨酸富集环上Tyr34、αC螺旋上Thr66形成了极性键;光甘草定还与ERK2中β1折叠上Glu31、β2折叠上Gly35、β3-折叠和αC螺旋之间的无规则卷曲区域的Ser55、Ile54和Pro56、αC螺旋上的Arg65、β2L0上的Gln15有许多疏水作用(图9-B)。光甘草定与ERK2的β-折叠(β1~β3)、αC螺旋及甘氨酸富集环上氨基酸残基的相互作用在空间上阻止了ATP与激酶的结合。因此,光甘草定可以被归类为ERK2的一种I1/2型抑制剂。
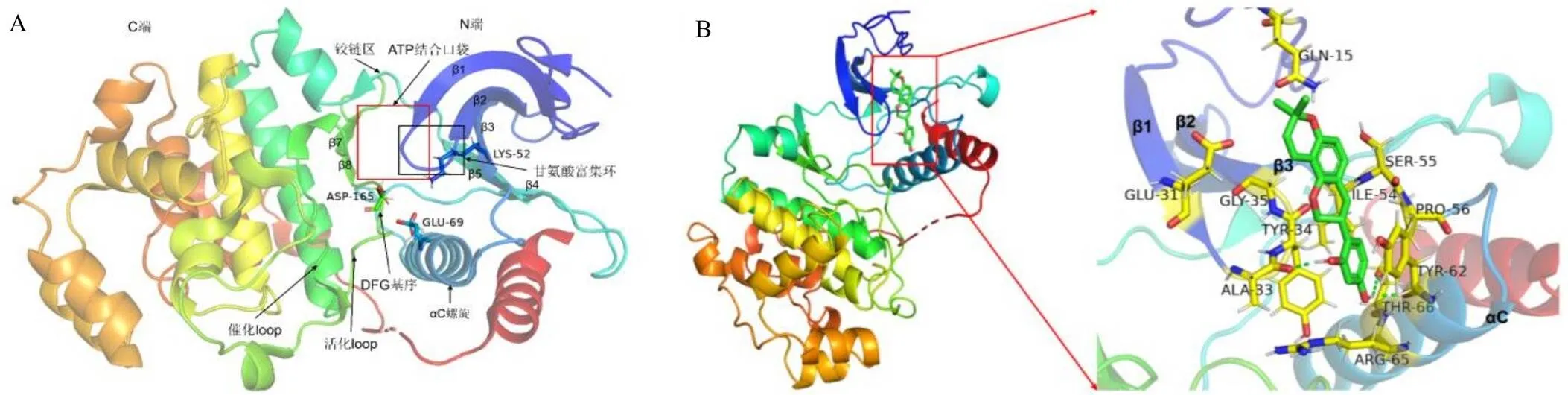
图9 ERK2结构图 (A)及光甘草定与ERK2的分子对接图 (B)
4 讨论
光甘草定具有显著降低IR-HepG2细胞肝糖产生的能力。肝糖的产生是糖酵解、糖原合成与分解、糖异生过程的等过程的净结果,对血糖调节起得重要作用。糖原在进食条件下是肝脏中葡萄糖的一种重要储存形式;在禁食条件下,肝糖原分解产生的游离葡萄糖释放到血液循环中,可随时为大脑和其他神经组织提供葡萄糖。伴随禁食时间的延长,糖异生逐渐取代糖原分解,成为肝脏葡萄糖生成的主途径。例如在禁食10 h后,糖原分解只占肝脏葡萄糖总产量的30%,糖异生占肝脏葡萄糖总产量的70%。糖异生也是T2DM患者空腹肝葡萄糖生成增加的主要原因[22]。发生IR时,肝脏的葡萄糖代谢失衡,包括糖酵解和糖原合成能力减弱、肝糖原的分解和糖异生加强,使得肝糖输出增加,最终造成肝脏调节血糖的能力减弱,血糖代谢出现紊乱。本研究表明,光甘草定通过上调PK、GCK和GS的活性,使细胞的糖酵解和糖原合成能力显著增强;通过下调PEPCK和G6Pase的活性,显著减弱了细胞内糖异生的能力,降低肝糖的产生,进而减少肝糖的输出。
光甘草定显著提高了IR-HepG2细胞的葡萄糖摄取能力。GLUT4是葡萄糖转运蛋白中独特的亚型,在介导胰岛素依赖性葡萄糖摄取和维持葡萄糖平衡方面发挥着重要作用[30]。GLUT4主要在脂肪细胞、骨骼肌和心肌细胞中表达,当摄入碳水化合物食物后,血液循环中的胰岛素水平上升;在胰岛素作用下,细胞内的GLUT4转位到质膜上,从而增加这些细胞的葡萄糖摄取和代谢,防止血糖水平的长期升高。GLUT4向质膜转移的缺陷被称为外周胰岛素抵抗。虽然目前认为GLUT2是肝细胞中主要的葡萄糖转运体,分别在进食和禁食状态下参与肝细胞葡萄糖的摄取和释放[31]。但是,GLUT4也在肝脏中表达[31],而且越来越多的体内外研究[17,32-35]表明,肝脏中的GLUT4通过与脂肪细胞、骨骼肌和心肌细胞中的GLUT4相同的机制,即通过胰岛素调节的转位参与肝细胞对血液中的葡萄糖的摄取。而当肝脏发生IR时,GLUT4也同样表现出易位损伤。GLUT4的易位损伤或修复与肝细胞摄取葡萄糖的能力密切相关。本研究结果显示,在高胰岛素诱导的IR-HepG2细胞中,GLUT4从细胞质向细胞膜的迁移受到了显著抑制,而光甘草定处理后质膜中GLUT4的表达显著提高,而细胞质中GLUT4的表达显著降低,表明光甘草定较好修复了IR-HepG2细胞的GLUT4的易位损伤。GLUT4在肝脏中的表达和功能以及对调节全身代谢方面的作用仍需进一步深入探讨。
PI3K/Akt途径是胰岛素信号转导的主要途径,在胰岛素的靶器官中负责调节葡萄糖摄取和代谢,Akt是该途径中的关键信号节点之一。在肝细胞中,胰岛素与细胞表面胰岛素受体特异性结合后,使IRS活化,接着IRS通过SH2结构域招募PI3K的p85调节亚基促使PI3K的激活,然后PI3K磷酸化脂质磷脂酰肌醇-4,5-二磷酸(phosphati-dylinositol-4,5-bisphosphate,PIP2)产生磷脂酰肌醇-3,4,5-三磷酸(phosphati-dylinositol-3,4,5-bisphosphate,PIP3);Akt通过其PH结构域与PIP3结合,促进上游激酶激活Akt。GSK-3β、FOXO1是Akt下游直接作用的2个靶点。GSK3β被Akt磷酸化而降低活性,致使GSK-3β的下游蛋白GS磷酸化水平下降而激活,进而促进糖原合成[36]。Akt使FOXO1发生磷酸化后,诱导FOXO1与14-3-3蛋白相互作用,使其发生核外排,致使FOXO1失去DNA结合能力和转录活性[37],导致PEPCK和G6Pase的表达降低,从而抑制糖异生[38]。当肝脏发生IR时,PI3K/Akt途径受损,Akt的激活受到了抑制,使其对GSK3β磷酸化的能力减弱,致使GSK3β通过磷酸化GS使其失活,减少了糖原的合成[39]。同时,Akt对FOXO1磷酸化能力也被削弱,FOXO1直接与PEPCK和G6Pase靶DNA序列结合,增加它们在肝脏中的表达[40],促进了肝糖异生。在本研究中,光甘草定显著上调了Akt的磷酸化水平,致使GSK3β和FOXO1的磷酸化水平也显著提高。前者导致GS的活性提高,IR-HepG2细胞的糖原含量也因此显著增加;后者导致PEPCK和G6Pase的活性显著降低,IR-HepG2细胞的葡萄糖生成量也因此显著下降。当加入PI3K抑制剂LY294002后,光甘草定对Akt、GSK3β和FOXO1磷酸化水平的上调作用被显著逆转。上述研究结果表明光甘草定改善IR的能力是PI3K/Akt途径依赖性的。
几个关键的丝氨酸/苏氨酸激酶通过靶向胰岛素信号传导PI3K/Akt途径的关键成分而成为重要的负向调节因子[15,41]。其中,ERK对IRS的丝氨酸磷酸化与IR密切相关[17]。过度激活的ERK信号通路对PI3K/Akt通路起负调节作用。在IR状态下,ERK激活后可以使IRS-1在Ser307等多个位点发生磷酸化[15],致使IRS-1与胰岛素受体的结合受到抑制[42],从而导致PI3K/Akt信号通路损伤和胰岛素刺激的葡萄糖利用受损。因此,抑制ERK途径可能成为治疗糖尿病的又一有效手段。在db/db小鼠[21,43]、KKAy小鼠[44]等动物模型和3T3-L1脂肪细胞[21,43]、骨骼肌细胞[16]中发现,使用抑制剂(PD184352、U0126、PD98059)或早期反应转录因子-1(early growth response-1,Egr-1)抑制ERK途径后,可改善小鼠和细胞的胰岛素敏感性及IR症状。近年来的研究表明,HepG2细胞的IR可通过核桃衍生肽[17]、姜黄素及其代谢产物[45]抑制ERK途径得到显著改善。本研究发现,高胰岛素诱导的IR-HepG2细胞的ERK和IRS-1的磷酸化水平显著升高,而这种升高分别被ERK通路的专一性抑制剂PD98059和光甘草定显著抑制。光甘草定与ERK2分子对接的结果进一步表明,光甘草定通过氢键、极性键、疏水相互作用等作用力稳定结合在非活性状态且具有DFG-in构象的ERK2的“Front cleft”和“Gate area”位点,在空间上阻止了ATP与ERK2的结合。上述结果表明,光甘草定可作为ERK2的一种I1/2型抑制剂,通过抑制IR-HepG2细胞中ERK/ IRS-1途径的激活,修复PI3K/Akt途径的损伤。
综上所述,光甘草定可作为ERK的I1/2型抑制剂,通过抑制IR-HepG2细胞的ERK/IRS-1通路、激活PI3K/Akt信号通路,促进GLUT4向质膜的易位,提高IR-HepG2细胞对葡萄糖的摄取量;增强或抑制葡萄糖代谢关键酶的活性,促进IR-HepG2细胞的糖原合成和糖酵解,抑制糖异生;进而提高IR-HepG2细胞对外源葡萄糖的利用、减少肝糖的产生和外排,修复葡萄糖的代谢紊乱,缓解IR症状。
利益冲突 所有作者均声明不存在利益冲突
[1] Virally M, Blicklé J F, Girard J,. Type 2 diabetes mellitus: Epidemiology, pathophysiology, unmet needs and therapeutical perspectives [J]., 2007, 33(4): 231-244.
[2] Dongiovanni P, Rametta R, Meroni M,. The role of insulin resistance in nonalcoholic steatohepatitis and liver disease development: A potential therapeutic target? [J]., 2016, 10(2): 229-242.
[3] Das S K, Elbein S C. The genetic basis of type 2 diabetes [J]., 2006, 2(4): 100-131.
[4] Simmler C, Pauli G F, Chen S N. Phytochemistry and biological properties of glabridin [J]., 2013, 90: 160-184.
[5] Yehuda I, Madar Z, Leikin-Frenkel A,. Glabridin, an isoflavan from licorice root, upregulates paraoxonase 2 expression under hyperglycemia and protects it from oxidation [J]., 2016, 60(2): 287-299.
[6] Wu F H, Jin Z G, Jin J. Hypoglycemic effects of glabridin, a polyphenolic flavonoid from licorice, in an animal model of diabetes mellitus [J]., 2013, 7(4): 1278-1282.
[7] Sawada K, Yamashita Y, Zhang T S,. Glabridin induces glucose uptake via the AMP-activated protein kinase pathway in muscle cells [J]., 2014, 393(1/2): 99-108.
[8] Choi E M, Suh K S, Kim Y J,. Glabridin alleviates the toxic effects of methylglyoxal on osteoblastic MC3T3-E1 cells by increasing expression of the glyoxalase system and Nrf2/HO-1 signaling and protecting mitochondrial function [J]., 2016, 64(1): 226-235.
[9] Yehuda I, Madar Z, Szuchman-Sapir A,. Glabridin, a phytoestrogen from licorice root, up-regulates manganese superoxide dismutase, catalase and paraoxonase 2 under glucose stress [J]., 2011, 25(5): 659-667.
[10] Yehuda I, Madar Z, Leikin-Frenkel A,. Glabridin, an isoflavan from licorice root, downregulates iNOS expression and activity under high-glucose stress and inflammation [J]., 2015, 59(6): 1041-1052.
[11] Choi E M, Suh K S, Jung W W,. Glabridin attenuates antiadipogenic activity induced by 2,3,7,8-tetrachlorodibenzo--dioxin in murine 3T3-L1 adipocytes [J]., 2018, 38(11): 1426-1436.
[12] Guo Y J, Pan W W, Liu S B,. ERK/MAPK signalling pathway and tumorigenesis [J]., 2020, 19(3): 1997-2007.
[13] Lawan A, Bennett A M. Mitogen-activated protein kinase regulation in hepatic metabolism [J]., 2017, 28(12): 868-878.
[14] Schultze S M, Hemmings B A, Niessen M,. PI3K/AKT, MAPK and AMPK signalling: Protein kinases in glucose homeostasis [J]., 2012, 14: e1.
[15] Tanti J F, Jager J. Cellular mechanisms of insulin resistance: Role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation [J]., 2009, 9(6): 753-762.
[16] Bouzakri K, Roques M, Gual P,. Reduced activation of phosphatidylinositol-3 kinase and increased serine 636 phosphorylation of insulin receptor substrate-1 in primary culture of skeletal muscle cells from patients with type 2 diabetes [J]., 2003, 52(6): 1319-1325.
[17] Wang J, Wu T, Fang L,. Peptides from walnut (Maxim.) protect hepatic HepG2 cells from high glucose-induced insulin resistance and oxidative stress [J]., 2020, 11(9): 8112-8121.
[18] Artunc F, Schleicher E, Weigert C,. The impact of insulin resistance on the kidney and vasculature [J]., 2016, 12(12): 721-737.
[19] Biddinger S B, Kahn C R. From mice to men: Insights into the insulin resistance syndromes [J]., 2006, 68: 123-158.
[20] Cargnello M, Roux P P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases [J]., 2011, 75(1): 50-83.
[21] Yu X, Shen N, Zhang M L,. Egr-1 decreases adipocyte insulin sensitivity by tilting PI3K/Akt and MAPK signal balance in mice [J]., 2011, 30(18): 3754-3765.
[22] Magnusson I, Rothman D L, Katz L D,. Increased rate of gluconeogenesis in type II diabetes mellitus. A13C nuclear magnetic resonance study [J]., 1992, 90(4): 1323-1327.
[23] Huang S H, Czech M P. The GLUT4 glucose transporter [J]., 2007, 5(4): 237-252.
[24] Roskoski R Jr. Targeting ERK1/2 protein-serine/threonine kinases in human cancers [J]., 2019, 142: 151-168.
[25] Roskoski R Jr. ERK1/2 MAP kinases: Structure, function, and regulation [J]., 2012, 66(2): 105-143.
[26] Dar A C, Shokat K M. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling [J]., 2011, 80: 769-795.
[27] Zuccotto F, Ardini E, Casale E,. Through the “gatekeeper door”: Exploiting the active kinase conformation [J]., 2010, 53(7): 2681-2694.
[28] Liao J J L. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors [J]., 2007, 50(3): 409-424.
[29] van Linden O P J, Kooistra A J, Leurs R,. KLIFS: A knowledge-based structural database to navigate kinase-ligand interaction space [J]., 2014, 57(2): 249-277.
[30] Harriet Wallberg-Henriksson. GLUT4: A key player regulating glucose homeostasis? Insights from transgenic and knockout mice [J]., 2001, 18(3): 205-211.
[31] Karim S, Adams D H, Lalor P F. Hepatic expression and cellular distribution of the glucose transporter family [J]., 2012, 18(46): 6771-6781.
[32] Zhang Y, Yan L S, Ding Y,.(Wall.) Meisn. water extract ameliorates palmitate induced insulin resistance by regulating IRS1/GSK3β/FoxO1 signaling pathway in human HepG2 hepatocytes [J]., 2020, 10: 1666.
[33] Garabadu D, Krishnamurthy S. Metformin attenuates hepatic insulin resistance in type-2 diabetic rats through PI3K/Akt/GLUT-4 signalling independent to bicuculline-sensitive GABAA receptor stimulation [J]., 2017, 55(1): 722-728.
[34] Li W Q, Wu L, Sun Q,. MicroRNA-191 blocking the translocation of GLUT4 is involved in arsenite-induced hepatic insulin resistance through inhibiting the IRS1/AKT pathway [J]., 2021, 215: 112130.
[35] Zhang W N, Su R N, Gong L L,. Structural characterization andhypoglycemic activity of a glucan fromSalisb. seeds [J]., 2019, 209: 363-371.
[36] Samuel V T, Shulman G I. Mechanisms for insulin resistance: Common threads and missing links [J]., 2012, 148(5): 852-871.
[37] Brownawell A M, Kops G J, Macara I G,. Inhibition of nuclear import by protein kinase B (Akt) regulates the subcellular distribution and activity of the forkhead transcription factor AFX [J]., 2001, 21(10): 3534-3546.
[38] Klover P J, Mooney R A. Hepatocytes: Critical for glucose homeostasis [J]., 2004, 36(5): 753-758.
[39] Srivastava A K, Pandey S K. Potential mechanism(s) involved in the regulation of glycogen synthesis by insulin [J]., 1998, 182(1/2): 135-141.
[40] Sekine K, Chen Y R, Kojima N,. Foxo1 links insulin signaling to C/EBPalpha and regulates gluconeogenesis during liver development [J]., 2007, 26(15): 3607-3615.
[41] Taniguchi C M, Emanuelli B, Kahn C R. Critical nodes in signalling pathways: Insights into insulin action [J]., 2006, 7(2): 85-96.
[42] Eck M J, Dhe-Paganon S, Trüb T,. Structure of the IRS-1 PTB domain bound to the juxtamembrane region of the insulin receptor [J]., 1996, 85(5): 695-705.
[43] Jager J, Grémeaux T, Cormont M,. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression [J]., 2007, 148(1): 241-251.
[44] Ozaki K I, Awazu M, Tamiya M,. Targeting the ERK signaling pathway as a potential treatment for insulin resistance and type 2 diabetes [J]., 2016, 310(8): E643-E651.
[45] Li P, Ding L Q, Cao S J,. Curcumin metabolites contribute to the effect of curcumin on ameliorating insulin sensitivity in high-glucose-induced insulin-resistant HepG2 cells [J]., 2020, 259: 113015.
Glabridin ameliorates insulin resistance by regulating ERK/IRS-1 and PI3K/Akt signaling pathways in HepG2 cells
LI De-feng, FAN Jin-ling, DU Lin, REN Guo-yan
College of Food and Bioengineering, Henan University of Science and Technology, Luoyang 471023, China
To explore the effect and mechanism of glabridin on ameliorating insulin resistance (IR) in hepatocytes.IR model was established by high insulin-induced HepG2 cells. The cells were evaluated for glucose consumption and production by glucose oxidase assay; The glucose consumption and production of cells were detected by glucose oxidase method; Glucose uptake was detected by fluorescence method; The content of glycogen was detected by anthrone method; The activities of key enzymes in glucose metabolism was detected by ELISA; Western blotting was used to detect phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt), extracellular regulated protein kinase/insulin receptor substrate-1 (ERK/IRS-1) signaling pathway related protein and glucose transporter 4 (GLUT4) expressions. Molecular docking technique was used to study the interaction between glabridin and ERK molecules.Glabridin significantly increased the glucose consumption and uptake of IR-HepG2 cells (< 0.05); Glycogen synthesis and glycolysis of IR-HepG2 cells were promoted by significantly increasing the activities of glycogen synthase (GS), glucokinase (GCK) and pyruvate kinase (PK) (< 0.05, 0.01); The activities of phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) were significantly decreased (< 0.05), and gluconeogenesis of IR-HepG2 cells was inhibited. After IR-HepG2 cells were treated with glabridin, phosphorylation levels of Akt, glycogen synthase kinase-3β (GSK-3β) and forkhead boxing protein O1 (FOXO1) were significantly restored (< 0.01), and this effect was reversed by PI3K inhibitor LY294002 (< 0.01). Meanwhile, glabridin significantly promoted the translocation of GLUT4 to plasma membrane (< 0.01). Glabridin significantly reduced the phosphorylation levels of ERK and IRS in IR-HepG2 cells (< 0.01), and could be used as I1/2inhibitor of ERK.Glabridin can repair the sugar metabolism disorder of IR-HepG2 cells and relieve IR symptoms by inhibiting ERK/IRS-1 pathway and activating PI3K/Akt signaling pathway.
insulin resistance; ERK/IRS-1; PI3K/Akt; glabridin; HepG2 cells; glucose metabolism; glucose uptake
R285.5
A
0253 - 2670(2022)24 - 7751 - 12
10.7501/j.issn.0253-2670.2022.24.013
2022-10-08
国家自然科学基金面上项目(31571800)
李德锋(1997—),男,硕士研究生,研究方向为天然产物组分与功能食品。E-mail: 1659628204@qq.cm
樊金玲(1973—),女,教授,博士生导师,研究方向为天然产物组分与功能食品。E-mail: fanjinling@haust.edu.cn
[责任编辑 李亚楠]
