右美托咪定联合七氟醚对糖尿病小鼠心肌缺血/再灌注损伤的保护作用
2022-12-28肖志博葛树胜吴小精尹顺花
金 辉,肖志博,葛树胜,吴小精,尹顺花,李 媛
(1.海南医学院第一附属医院麻醉科,海南 海口 570100;2.海南省妇女儿童医学中心麻醉科,海南 海口 570100)
近年来,糖尿病的发病率逐年上升,并呈年轻化趋势,已成为严重威胁人类健康的全球性公共卫生问题。2019年全球65岁及以上的人群中约有193万人患有糖尿病,预计2045年将达到2 762万[1]。心血管疾病,特别是急性冠状动脉综合征是糖尿病患者的重要合并症,其危害远大于糖尿病本身。心肌缺血/再灌注损伤(myocardial ischemia/reperfusion injury,MI/RI)是临床常见的病理生理过程,心肌发生缺血后,应及时行血液再灌注恢复血液供应以防止心肌死亡,但该过程也会对心肌造成二次损伤,这种现象被称为MI/RI[2]。研究发现,糖尿病患者心肌缺血的发生率比非糖尿病患者高1.45~2.99倍,这对糖尿病患者来说是一个严重的健康威胁[3]。而且长期患有糖尿病可引发血管内皮功能障碍,这将加剧缺血/再灌注(ischemia/reperfusion,I/R)引起的心肌损伤[4-5]。
七氟醚(sevoflurane,Sev)是一种广泛用于临床和基础研究的吸入麻醉剂。研究证实,七氟醚-后处理(sevoflurane-post conditioning,Sev-postC)可以有效减轻健康心肌的I/R损伤[6-7];而高血糖可抵消Sev-postC的心肌保护作用,但其具体机制尚不清楚。有研究显示,巨噬细胞迁移抑制因子(macrophage migration inhibitory factor,MIF)在MI/RI过程中发挥着重要作用,而高血糖能够抑制MIF的表达[8-9]。右美托咪定(dexmedetomidine,Dex)是一种高度选择性的α2受体激动剂,通常在临床中用作镇静剂。研究证实,Dex能够有效保护心肌免于I/R损伤,其保护机制包括上调缺氧诱导因子-1α(hypoxia inducible factor 1 subunit alpha,HIF-1α)的表达[10]。HIF-1α在心肌缺血过程中被显著诱导表达,其在心肌缺血/缺氧过程中是启动内源性心肌保护机制的起始因子,是防御MI/RI的主要调节因子[11]。而MIF是HIF-1α的下游效应因子,心肌组织中HIF-1α与MIF的表达密切相关[9,11]。因此,本研究拟通过体外研究和在体构建糖尿病小鼠MI/RI模型,探讨Dex联合Sev-postC对糖尿病小鼠MI/RI的保护作用及其分子机制。
1 材料与方法
1.1 实验材料
6~8周龄SPF级雄性C57 BL/KS m/m(Lepr-WT/WT)小鼠15只,体质量(23±2)g;6~8周龄SPF级雄性C57 BL/KS db/db(Lepr-KO/KO)2型糖尿病小鼠20只,体质量(41±3)g,购自海南药物研究所有限责任公司;H9c2大鼠胚胎心肌细胞购自武汉普诺赛生物;FBS(批号:10091155),低葡萄糖DMEM(批号:11966025),高葡萄糖DMEM(葡萄糖35 μmol/L;批号:12100061)购自美国Gibco;Sev(批号:SF-2785016),Dex(批号:YT-1732234),戊巴比妥钠(批号:PZ-2437186)购自北京阳光生物;乳酸脱氢酶(lactate dehydrogenase,LDH)、肌酸激酶同工酶MB(creative kinase isoenzyme MB,CK-MB)ELISA试剂盒(批号:CSB-E11723m、CSB-E14404m),超氧化物歧化酶(superoxide dismutase,SOD)活性测定试剂盒(批号:CSB-E08556m)购自武汉华美;丙二醛(malondialdehyde,MDA)测定试剂盒(批号:S0131S),CCK-8细胞活力测定试剂盒(批号:C0037)购自上海碧云天;BCA蛋白定量试剂盒(批号:71285-3),ECL化学发光底物(批号:WBULS0500),蛋白酶抑制剂混合物(批号:P2714)购自美国Merck-Millipore;RIPA裂解液(批号:89901)购自美国Thermo Fisher;兔抗HIF-1α单抗(批号:ab179483),兔MIF单抗(批号:ab187064),山羊抗兔IgG(H+L)HRP二抗(批号:ab3721)购自美国Abcam;酶标仪(型号:800TS)购自美国BioTek公司;光学显微镜(型号:Ci-E)购自日本Nikon。
1.2 体外和在体实验分组
体外实验分组:①LG-Control组,低糖(LG)培养条件下培养H9c2心肌细胞;②LG-H/R组,低糖培养条件下对H9c2心肌细胞进行缺氧/复氧(hypoxia/reoxygenation,H/R)处理;③LG-Sev组,低糖培养条件下对H9c2心肌细胞进行H/R处理后,加入Sev处理细胞;④HG-Control组,高糖(HG)培养条件下培养H9c2心肌细胞;⑤HG-H/R组,高糖培养条件下对H9c2心肌细胞进行H/R处理后,在常氧条件下继续培养12 h;⑥HG-Sev组,高糖培养条件下对H9c2心肌细胞进行H/R处理后,加入Sev处理细胞,在常氧条件下继续培养6 h;⑦HG-Sev+Dex组,高糖培养条件下对H9c2心肌细胞进行H/R处理后,在对细胞进行Sev处理的同时加入Dex处理细胞,然后收获细胞。
在体实验分组(n=5):①Sham组,正常小鼠MI/RI模型假手术;②MI/RI组,正常小鼠构建MI/RI模型;③Sev-postC组,对正常小鼠构建MI/RI模型时,在再灌注前持续吸入2.5% Sev 15 min[12];④DM-Sham组,糖尿病小鼠MI/RI模型假手术;⑤DM-MI/RI组,糖尿病小鼠构建MI/RI模型;⑥DM-Sev-postC组,对糖尿病小鼠构建MI/RI模型时,在再灌注前持续吸入2.5% Sev 15 min[12];⑦DM-Sev-postC+Dex组,对糖尿病小鼠构建MI/RI模型时,在再灌注前持续吸入 2.5% Sev 15 min,再灌注结束后腹腔注射10 μg/kg Dex[13]。
1.3 细胞培养与药物处理
高糖培养:H9c2心肌细胞置于含10%FBS的高葡萄糖DMEM培养液中,培养于5%CO2、37 ℃恒温孵育箱中。低糖培养:在高糖培养条件下细胞生长汇合度达90%,且生长状态良好时,消化细胞,并以4×106个/皿的密度接种于6孔板中,继续培养24 h;第2天当细胞生长汇合度达80%时,弃去细胞培养液,贴壁细胞用37 ℃预热的PBS洗涤3次后,加入无血清低葡萄糖DMEM培养液继续培养。Sev处理:向细胞中加入3.4%的Sev处理6 h[14]。Sev+Dex处理:向细胞中加入3.4%的Sev处理6 h,同时加入1 μmol/L的Dex处理12 h[15]。
1.4 体外诱导H9c2心肌细胞H/R损伤模型
H/R损伤模型:取2×106个细胞接种于6孔板中,待细胞生长汇合度达50%,适应性贴壁后,将细胞培养液更换为无血清DMEM(高糖或低糖)培养液,置于含0.1%O2、5%CO2和95%N2的低氧室中,在37 ℃恒温、湿润的条件下缺氧培养7 h。随后更换为含10%FBS的DMEM(高糖或低糖)完全培养液,置于常氧条件下正常培养2 h,收获细胞检测下游指标。Control组细胞于含10%FBS的DMEM(高糖或低糖)完全培养液的常氧条件下培养。
1.5 MI/RI小鼠模型
MI/RI造模前,对小鼠进行为期1周的适应性饲养。每4只小鼠1笼,饲养于SPF级鼠房,12 h光照/12 h黑暗周期循环,可自由进食和饮水。
整个手术期间都将小鼠放置在30 ℃恒温加热垫上。于小鼠腹腔注射2%戊巴比妥钠(60 mg/kg)进行麻醉,监测小鼠肢体张力、自主呼吸和心率。麻醉后进行气管插管,连接到动物呼吸机进行机械通气。术前开始记录小鼠心电图。MI/RI组:小鼠开胸后,暴露心脏,用7/0无创缝合线结扎冠状动脉左前降支;通过心电图识别ST段,20 min后松开结扎线进行再灌注。Sham组:小鼠仅开胸,将7/0无创缝合线放置于冠状动脉左前降支部位,但并不进行结扎;手术后,继续监测小鼠心电图,缝合胸腔,并肌肉注射青霉素(400 kU/kg)。对于MI/RI组和Sham组小鼠,均在麻醉条件下[2%戊巴比妥(60 mg/kg)腹腔注射]采集小鼠腹主动脉血样。待小鼠苏醒后,继续进行再灌注 24 h,然后对小鼠进行安乐死[2%戊巴比妥(100 mg/kg)腹腔注射][13]。随后立即采集小鼠心脏样本,用于下游指标的检测。
1.6 心肌细胞存活能力测定
使用CCK-8细胞活力测定试剂盒测定心肌细胞存活能力。将H9c2心肌细胞接种于96孔板中,细胞贴壁后,按照各组的要求处理细胞。处理结束后,向每孔细胞中加入试剂盒自带的10% CCK-8溶液100 μL,在37 ℃培养箱中孵育2 h。然后使用BioTek酶标仪在450 nm波长下测量光密度值。细胞存活率(%)=各组细胞光密度值/LG-Control组细胞光密度值×100%。
1.7 HE染色
新鲜小鼠心脏首先浸没于10%中性福尔马林中,在4 ℃下固定48 h,然后进行石蜡包埋并切片(5 μm)。切片经2次二甲苯彻底脱蜡和梯度乙醇脱水,然后使用苏木精染色液染色5 min;分化液分化 3 s;切片复蓝约3 s。再用85%和95%乙醇脱水,每次 4 min。然后对切片进行伊红染色5 min;无水乙醇脱水3次,每次5 min;二甲苯透明化2次,每次 2 min;最后,中性树胶封片。在Nikon光学显微镜下观察和拍照。
1.8 ELISA检测
全血样先进行离心(3 000 ×g,5 min),获得血清样本。然后按照ELISA试剂盒说明书检测LDH和CK-MB水平。按照成品试剂盒操作说明书检测SOD活性和MDA水平。使用BCA蛋白定量试剂盒检测组织或细胞总蛋白浓度。使用BioTek酶标仪在450 nm波长处读取吸光值。
1.9 Western blot检测
于心脏组织或心肌细胞中加入含有蛋白酶抑制剂混合物的RIPA裂解液,在液氮中研磨组织为组织匀浆;或使用超声破碎机在冰浴中破碎心肌细胞。使用BCA蛋白定量试剂盒检测总蛋白浓度。取10 μg总蛋白/样本进行SDS-PAGE凝胶电泳;采用半干法电转印(恒流100 mA),将凝胶中的蛋白电转印至PVDF膜上。使用含10%脱脂奶粉的TBS-T对PVDF膜进行封闭,室温条件下封闭1 h。然后将PVDF膜与兔抗HIF-1α单抗(稀释度1∶1 000)或兔MIF单抗(稀释度1∶1 000)一抗于4 ℃共同孵育过夜;第2天使用TBS-T洗膜3次,每次5 min,然后将PVDF膜与山羊抗兔IgG(H+L)HRP二抗(稀释度1∶10 000)于37 ℃共同孵育1 h。使用ECL化学发光底物进行化学发光和条带显色。
1.10 统计学分析
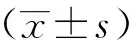
2 结果
2.1 高糖抑制MIF表达抵消Sev对心肌细胞H/R损伤的保护作用
与LG-Control组相比,LG-H/R组细胞存活能力显著降低(P<0.01);与LG-H/R组相比,LG-Sev组细胞存活能力显著升高(P<0.01);与HG-Control组相比,HG-H/R组细胞存活能力显著降低(P<0.01);HG-Sev组与HG-H/R组细胞存活能力比较差异无统计学意义(P>0.05),见图1a。与LG-Control组相比,LG-H/R组细胞SOD水平显著降低(P<0.01),LDH、CK-MB、MDA水平均显著升高(P<0.01);与LG-H/R组相比,LG-Sev组SOD水平显著上升(P<0.01),LDH、CK-MB、MDA水平均显著降低(P<0.01)。与HG-Control组相比,HG-H/R组细胞SOD水平显著降低(P<0.01),LDH、CK-MB、MDA水平均显著升高(P<0.01);HG-Sev组与HG-H/R组SOD、LDH、CK-MB、MDA水平比较差异均无统计学意义(P>0.05),见图1b~e。与LG-Control组相比,LG-H/R组细胞HIF-1α表达显著升高(P<0.01);与LG-H/R组相比,LG-Sev组HIF-1α表达显著降低(P<0.01)。与HG-Control组相比,HG-H/R组细胞HIF-1α表达显著升高(P<0.01);而HG-H/R组与HG-Sev组HIF-1α表达比较差异无统计学意义(P>0.05)。与LG-Control组相比,LG-H/R组MIF表达显著上调(P<0.01);与LG-H/R组相比,LG-Sev组MIF表达显著下调(P<0.01)。HG-Control组、HG-H/R组、HG-Sev组MIF表达比较差异均无统计学意义(P>0.05)。与LG-H/R组相比,HG-H/R组细胞MIF表达显著降低(P<0.01),见图1f。

a:细胞存活率;b:LDH水平;c:CK-MB水平;d:SOD水平;e:MDA水平;f:HIF-1α和MIF蛋白表达 *:P<0.05;#:P<0.01
2.2 Dex通过上调MIF增强Sev处理对心肌细胞H/R损伤的保护作用
与HG-Control组相比,HG-H/R组细胞存活能力显著降低(P<0.01);与HG-H/R组相比,HG-Sev组细胞存活能力无显著变化(P>0.05),而HG-Sev+Dex组细胞存活能力显著提高(P<0.01),见图2a。与HG-Control组相比,HG-H/R组细胞SOD显著降低(P<0.01),LDH、CK-MB、MDA显著升高(P<0.01);与HG-H/R相比,HG-Sev组LDH、CK-MB、MDA、SOD水平无显著变化(P>0.05),而HG-Sev+Dex组LDH、CK-MB、MDA水平显著降低(P<0.01),SOD水平升高(P<0.01),见图2b~e。与HG-Control组相比,HG-H/R组细胞HIF-1α的表达显著升高(P<0.01);与HG-H/R组相比,HG-Sev组HIF-1α的表达无显著变化(P>0.05),而HG-Sev+Dex组HIF-1α的表达显著下调(P<0.01)。与HG-Control组相比,HG-H/R组、HG-Sev组细胞MIF的表达无显著变化(P>0.05),HG-Sev+Dex组MIF的表达显著上调(P<0.01)。与HG-H/R组相比,HG-Sev组MIF的表达无显著变化(P>0.05),HG-Sev+Dex组MIF的表达显著上调(P<0.01),见图2f。

a:细胞存活率;b:LDH水平;c:CK-MB水平;d:SOD水平;e:MDA水平;f:HIF-1α和MIF蛋白表达 *:P<0.05;#:P<0.01
2.3 Dex通过上调MIF增强Sev-postC对糖尿病小鼠MI/RI的心肌保护作用
小鼠在体实验结果显示,与Sham组相比,MI/RI组、DM-Sham组小鼠心肌HIF-1α表达水平显著升高(P<0.01);与MI/RI组相比,Sev-postC组HIF-1α表达水平明显降低(P<0.05);与DM-Sham组相比,DM-MI/RI组HIF-1α表达水平明显升高(P<0.01);与DM-MI/RI组相比,DM-Sev-postC组HIF-1α表达水平无显著变化(P>0.05),DM-Sev-postC+Dex组HIF-1α表达水平明显下调(P<0.01)。与Sham组相比,MI/RI组MIF表达水平显著升高(P<0.01),DM-Sham组MIF表达水平显著降低(P<0.01);与MI/RI组相比,Sev-postC组MIF表达水平显著降低(P<0.01);与DM-MI/RI组相比,DM-Sham组和DM-Sev-postC组MIF表达水平无显著变化(P>0.05),而DM-Sev-postC+Dex组MIF表达水平显著升高(P<0.01),见图3a。HE染色结果显示,DM-Sham组小鼠心肌细胞排列整齐有序,心肌纤维无断裂及结构破坏;DM-MI/RI组小鼠心肌细胞排列杂乱,心肌纤维断裂和结构破坏严重;与DM-MI/RI组小鼠相比,DM-Sev-postC组小鼠心肌损伤无明显缓解,而DM-Sev-postC+Dex组小鼠心肌损伤明显缓解,心肌细胞排列较为整齐有序,心肌纤维无断裂,结构破坏不明显(图3b)。与DM-Sham组相比,DM-MI/RI组LDH、CK-MB和MDA水平显著升高(P<0.01),SOD水平显著降低(P<0.01);与DM-MI/RI组相比,DM-Sev-postC组LDH、CK-MB、MDA、SOD水平无显著变化(P>0.05),DM-Sev-postC+Dex组LDH、CK-MB和MDA水平明显降低(P<0.01),SOD水平明显升高(P<0.01),见图3c。

a:心肌组织中HIF-1α和MIF蛋白表达;b:心肌组织HE染色;c:心肌组织中LDH、CK-MB、SOD和MDA水平 *:P<0.05;#:P<0.01
3 讨论
对于ST段抬高型心肌梗死患者,通过直接经皮冠状动脉介入治疗(percutaneous coronary intervention,PCI)进行快速再灌注是目前限制梗死面积的最佳干预措施[16],然而缺血和再灌注会引起I/R损伤。在动物实验和临床研究中,氧气和血液供应恢复导致的再灌注损伤面积可占最终心肌梗死面积的50%[16]。
尽管及时再灌注,仍然会导致明显的心肌坏死,这种I/R损伤的程度是PCI后预后的主要决定因素。已有不同的心脏保护方法来限制ST段抬高型心肌梗死患者在直接PCI期间的梗死面积,其中通过向静脉或冠状动脉内注射药物或细胞已被证明是安全有效的,但仍需要进一步的研究以获得满意的临床结果。
多数临床前和临床数据表明,糖尿病是急性冠状动脉综合征患者的重要合并症;糖尿病患者的心脏最容易受到I/R损伤;有糖尿病存在的情况下,缺血预处理和药物等对心脏的保护作用会削弱。因此糖尿病患者发生急性心血管事件的可能性更大,而且心脏保护措施减少梗死面积的可能性更有限[16]。本研究也显示,高糖培养条件下对细胞进行H/R处理后,给予Sev治疗的效果没有低糖培养条件下对细胞进行相同处理时明显。
糖尿病患者对I/R损伤的易感性增加是由多种机制引发的,包括心肌细胞促存活信号通路的激活不足、心肌细胞线粒体能量代谢的改变、活性氧自由基的异常产生以及各种胞内和胞外位点抗氧化能力受损,而且持续的高血糖还会减少一氧化氮的产生和释放[16]。有研究显示,无论是在缺血前还是在再灌注前给予Dex,都能够提高肾细胞的存活能力,Dex通过抑制PI3K-Akt通路和ERK通路的磷酸化和活化,实现促缺血缺氧细胞存活能力的功效[17-19]。同时,Dex还能通过抑制缺氧细胞的氧化应激保护肾组织[20]。但是对于Dex作用于心肌细胞是否有类似的效果,目前尚不清楚。本研究结果显示,在高糖培养条件下,对心肌细胞仅给予Sev治疗无明显效果,而给予Sev+Dex后,可显著提高HG-H/R处理后心肌细胞的LDH、CK-MB、SOD和MDA水平;同时,还可降低HG-H/R处理后心肌细胞中LDH、CK-MB、SOD和MDA水平,从而减轻氧化应激。
HIF-1α是一种氧敏感转录因子,可通过转录激活200多个基因使生物体适应缺氧,被认为是缺氧和缺血信号转导激活的主要开关,其通过调节细胞内氧稳态、血管生成、心脏干细胞分化发挥保护作用[7]。研究发现HIF-1α可上调MIF的表达,而MIF可激活促存活信号通路(如AMPK磷酸化)以发挥心脏保护作用;缺血预处理通过MIF激活RISK和AMPK信号通路,减少心肌梗死面积和心肌细胞凋亡,抑制活性氧自由基产生和细胞的炎症浸润,维持内皮细胞功能,改善由MI/RI引起的心功能障碍,在心肌保护中发挥关键作用[8]。还有研究表明,MIF能抑制JNK介导的细胞凋亡,并通过其抗氧化能力在MI/RI期间发挥心肌保护作用[9]。但是MIF的表达是否受高血糖的影响仍未可知。本研究结果显示,MIF的表达在高糖条件下被抑制;同时,Sev-postC无法发挥心肌保护作用;在Sev-postC+Dex治疗后,能够上调MIF,并且发挥心肌保护作用。由此可以看出,高糖抑制了MIF的表达,Sev-postC对糖尿病小鼠的心肌保护作用消失,说明MIF很可能在其中发挥关键的作用。进一步研究发现,Dex在高血糖情况下依然能够上调MIF的表达,抵消高血糖抑制MIF表达的作用。对这一结果可能的解释有:①Dex能够稳定糖尿病小鼠的血糖波动[21];②Dex具有在高血糖情况下显著抑制MI/RI引起的氧化应激的能力[22]。后者在本研究结果中也得到了证实,Sev-postC+Dex处理能够显著抑制DM-MI/RI引起的氧化应激水平。
本研究初步证明了Dex联合Sev-postC能够明显缓解糖尿病小鼠心肌I/R引起的损伤。然而,本研究尚未对Dex通过何种途径提高MIF的表达进行深入探讨,Dex是否能够通过影响miRNAs发挥上调MIF保护心肌的作用,这是我们今后将要深入开展的研究方向,从而为深入理解Dex保护心肌免受I/R损伤的机制和临床上安全应用Dex进行针对性的治疗提供参考。
