新TMEM67基因突变致眼-脑-肝-肾综合征的诊断及家系研究*
2022-12-24杨灿金皎
杨灿, 金皎
(1.贵州医科大学附属医院 儿科血液专科, 贵州 贵阳 550001; 2.贵州医科大学 临床医学院, 贵州 贵阳 550001)
眼-脑-肝-肾综合征(eye-brain-liver-kidney syndrome),又名COACH综合征,是一种罕见的常染色体隐性遗传疾病,属于纤毛病中的一种,是指原发性纤毛缺陷会导致相关的多系统障碍畸形综合征[1];COACH综合征以先天性肝纤维化(congenital hepatic fibrosis,CHF)、小脑蚓部发育不全、少尿、共济失调及眼部结构缺损等为主要特征,其突变频率最高的基因是跨膜蛋白67(transmembrane protein 67,TMEM67)[2]。目前有报道认为COACH综合征是Joubert综合征的1个亚型,最核心的特征为CHF[3-4]。COACH综合征为罕见病,相关报道较少,导致临床医师对该病的识别及诊断困难,现报道本院诊断的具有新发突变的1个COACH综合征家系病例,以提高临床对该病的认识及诊断能力。
1 对象与方法
1.1 研究对象
1例COACH综合征的先证者及其家系,先证者要求符合COACH综合征的临床表现[3],家系成员为先证者的祖父母、外祖父母、父母、2位叔叔、2位舅舅、1位弟弟、3位表弟妹及3位堂兄妹。本研究获得医院伦理委员会审批[2022(005)]及家属知情同意。
1.2 研究方法
1.2.1先证者临床资料及其家系资料 收集先证者的出生史、个人史、既往史、家族史、体格检查、血常规、肝功能、肾功能、腹部超声、肝脏弹力纤维检测、眼底检查、光学相干断层成像(optical coherence tomography,OCT)检查及颅脑磁共振成像(magnetic resonance imaging,MRI)检查结果,收集先证者家系资料(包括病史和体格检查)。
1.2.2全外显子组捕获测序技术 取先证者及其3代内家系成员外周血各2 mL,采用基因组DNA提取试剂盒(康为世纪公司)提取先证者及其家系DNA[5];采用Covaris打断法将DNA打碎至100~700 bp片段,末端修复加“A”,放入基因扩增仪(polymerase chain reaction,PCR仪;美国Applied Biosystems公司)中扩增,得到文库DNA[6];经生物素标记的探针与文库 DNA 在一定条件下进行杂交,用链霉亲和素修饰的磁珠进行吸附,抓取目的基因[7];文库DNA运用“桥式”扩增反应加载到测序芯片 Flowcell 上,NextSeq 500仪器(美国Illumina公司)进行自动循环和成像,获完整核酸序列[8];运用Bwa、Samtools及Picard等软件对核酸序列进行分析。以上过程均由迈基诺公司技术人员完成。
2 结果
2.1 一般资料
先证者,男性,11岁,因“腹胀6年,腹痛3 d”就诊;无视物不清和模糊,5岁时开始出现腹胀,10岁时发现肝脾严重肿大并肝功能异常、全血细胞减少、智力发育较同龄儿落后;1岁4月开始说话,4岁会行走,学习成绩落后;双眼内聚、无鼻根低平,腹软、肝脾肿大、肝右肋下9.5 cm和剑突下6 cm、脾左肋下13 cm、质韧、边钝、光滑、无压痛,右足3、4趾并趾畸形,智力、运动发育迟缓;右眼视力0.3、左眼视力0.4,眼球不规则震颤,视盘发育异常、下方盘沿组织缺失,视盘血管走形异常、血管变细及视网膜色素异常(图1);母孕期及出生史无特殊,父母体健,非近亲结婚,先证者大舅幼年时因高热后出现智力发育落后,余家系无类似病例。
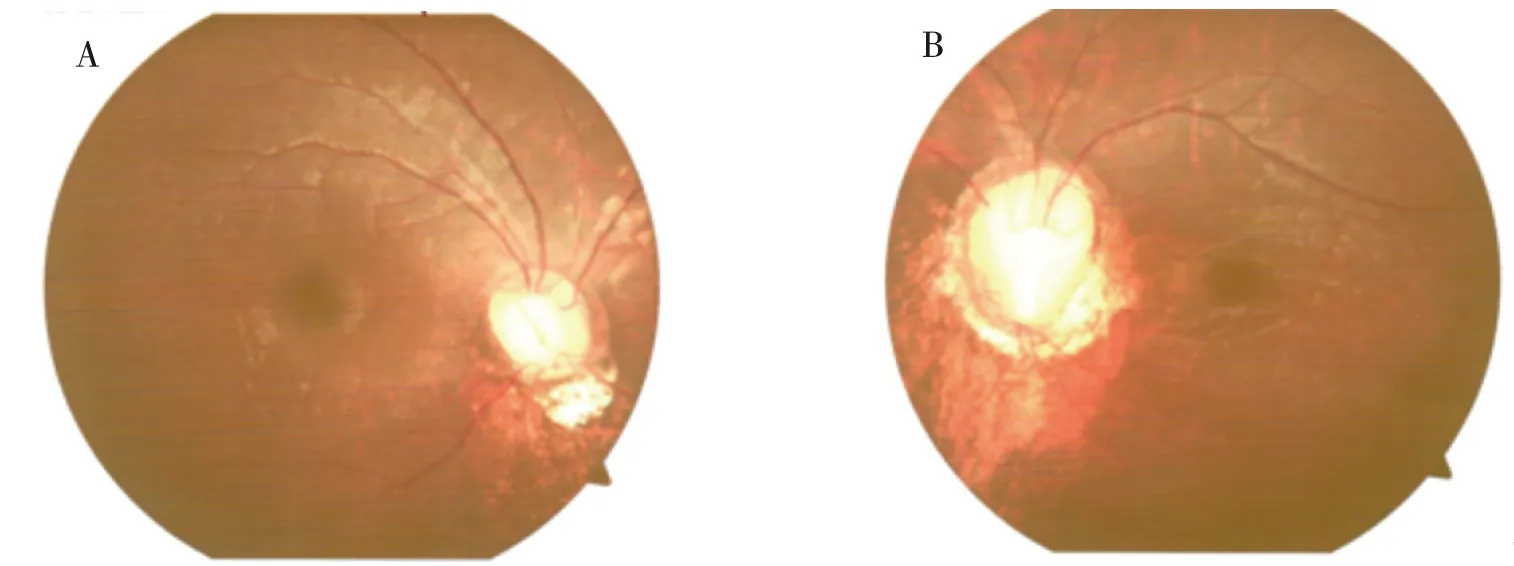
注:A、B分别为 左眼和右眼。图1 COACH综合征先证者的眼底检查结果Fig.1 Fundus examination results of the proband with COACH syndrome
2.2 实验室检查
血常规提示先证者3系减少,肝功能提示肝酶稍高,肾功能未见明显异常;腹部超声提示肝脾增大,肝脏回声减低,脾静脉增宽,肝门区迂曲走行静脉; FibroScan技术检查肝脏硬度55.1 kPa,肝脏纤维化等级F4; OCT检测为双眼黄斑中心凹以外视网膜外层结构缺失(图2);头颅MRI见“磨牙征”和“蝙蝠翼外观”表现(图3)。
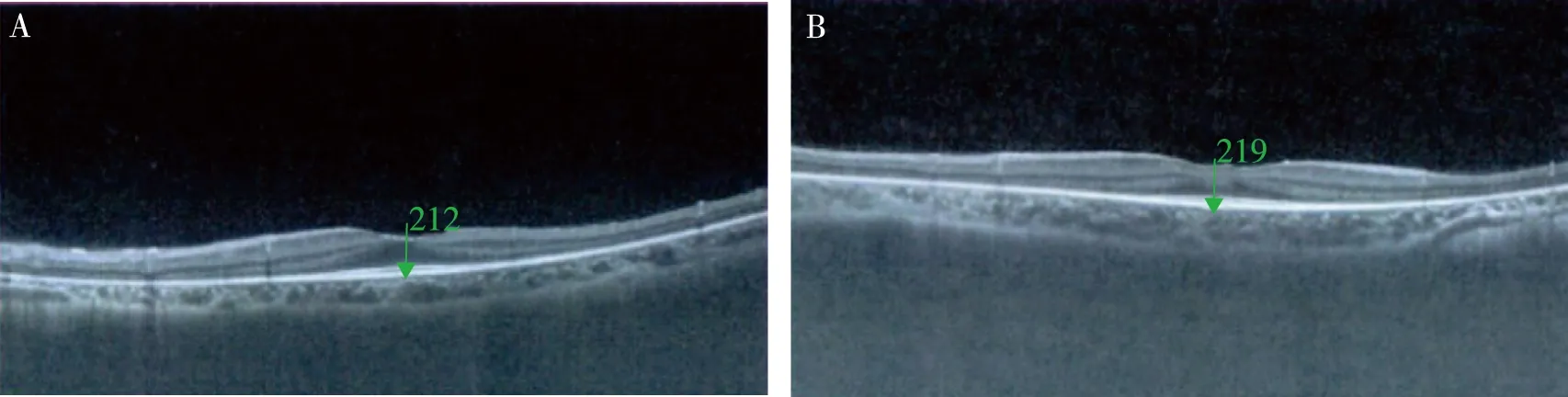
注:A、B分别为 左眼和右眼,绿色箭头表示黄斑中心凹厚度测量位置,绿色数字表示黄斑中心凹厚度(μm)。图2 COACH综合征先证者的眼部光学相干断层成像Fig.2 Optical Coherence Tomography findings of the proband with COACH syndrome

注:A、B、C分别为头颅MRI大脑中部水平面T2加权成像、矢状面T1加权成像、水平面T1加权成像;箭头分别指向“磨牙征”、增粗延长的小脑蚓部、“蝙蝠翼外观”。图3 COACH综合征先证者的头颅MRI检查结果Fig.3 Brain MRI findings of the proband with COACH syndrome
2.3 致病基因检测
先证者全外显子基因测序结果表明,检测出8号染色体(chr8)TMEM67 c.725A>G(p.N242S)和c.1288+1G>A(splicing)双重杂合突变,其中c.725A>G(p.N242S)突变为已报道的致病突变点,c.1288+1G>A(splicing)突变目前未被报道;经家系调查,c.725A>G(p.N242S)突变为父系遗传,c.1288+1G>A(splicing)突变为母系遗传,符合孟德尔遗传学定律(图4和图5);患儿大舅为Ⅱ代9号,携带c.1288+1G>A(splicing)突变。

注:Ⅰ、Ⅱ、Ⅲ分别表示一代家系、二代家系及三代家系,阿拉伯数字表示每代中的个体;□○分别表示正常男女,表示携带c.725A>G(p.N242S)突变,分别表示携带c.1288+1G>A(splicing)突变的男、女,■表示COACH综合征先证者。图4 COACH综合征先证者及其3代家系的致病基因遗传家系Fig.4 Genetic pedigree tree of pathogenic genes in the probands with COACH syndrome and the family members within three generations
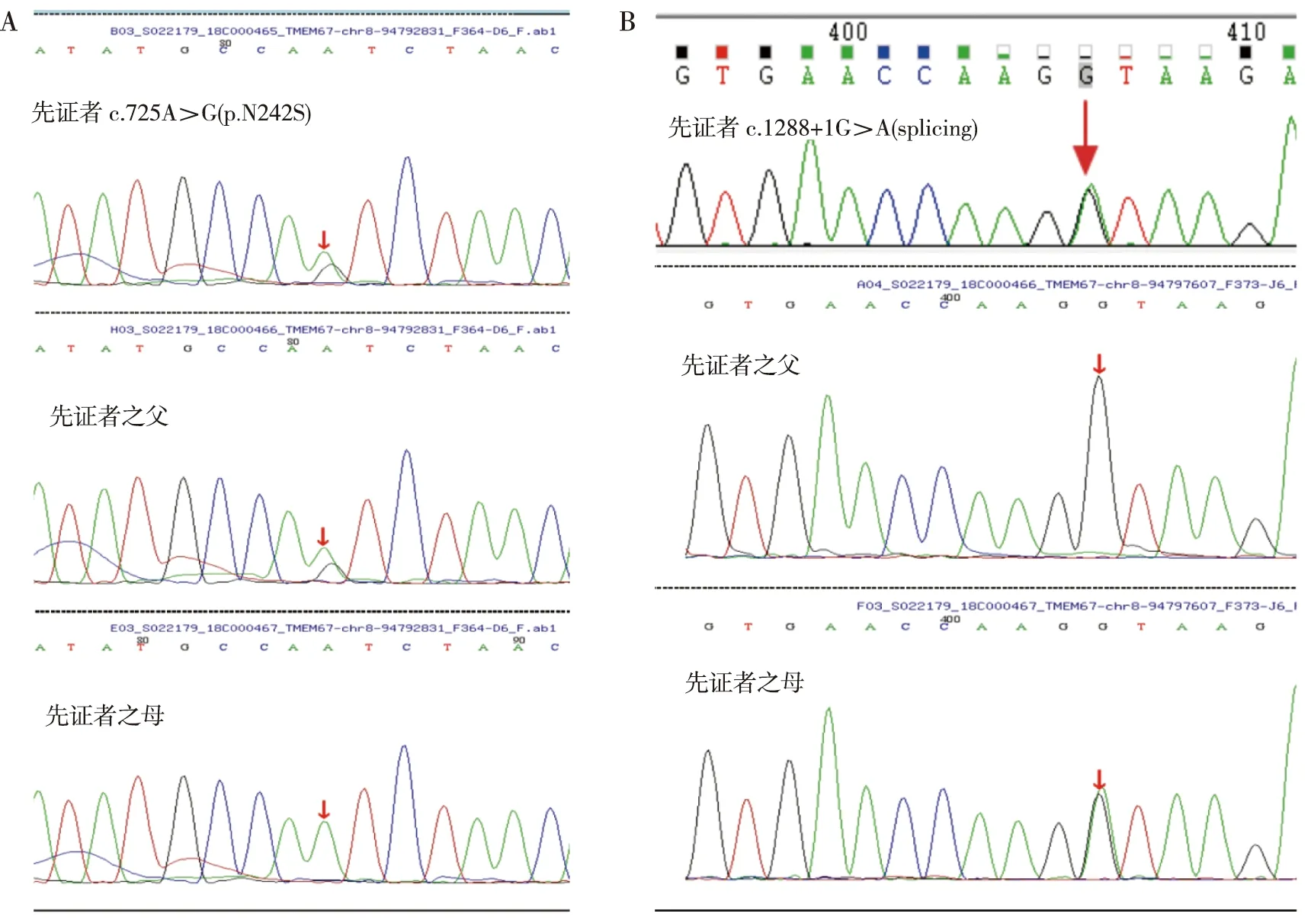
注:A、B分别为先证者c.725A>G(p.N242S)突变位点和c.1288+1G>A(splicing)突变位点的遗传来源;红色箭头表示突变的位点,颜色曲线表示不同的序列同源性。图5 COACH综合征患儿及其父母的TMEM67基因测序Fig.5 TMEM67 gene sequencing of the patient with COACH syndrome and his parents
3 讨论
COACH综合征是一种罕见的常染色体隐形遗传病,发病率低,国内外相关报道较少,且多为病例报告,无明显人种、性别及分布差异[2,9]。该病具有Joubert综合征基本的临床表现,如可有智力发育落后、眼部组织缺失、眼球震颤、并趾、肌张力异常[9],尤其是头颅MRI显示“磨牙征”或“蝙蝠翼”等表现[10-11],但COACH综合征最特征性的表现为CHF征象。国内外学者的研究均表明,COACH综合征患者均出现严重的智力发育落后及CHF,头颅MRI均发现典型的“磨牙征”或“蝙蝠翼征”[12-17]。多数临床研究结果显示,COACH综合征患者还会出现眼部症状,如视网膜营养不良、眼球震颤、眼组织缺失等症状[12,14-15,18]。本研究中COACH综合征患儿除了典型的CHF征象、智力发育落后、MRI呈现典型的“磨牙征”和“蝙蝠翼征”以外,还出现了眼部视网膜外层结构的缺损、并趾畸形等特征。
虽然生长发育迟缓、小脑蚓部结构缺失、CHF及眼部结构缺失等是COACH综合征普遍存在的特征,对其诊断具有高度指向性,但明确诊断仍需要结合基因分析。既往文献中报道了TMEM67、人性连锁视网膜色素变性GTP酶调节因子结合蛋白1 [human chain retinitis pigmentosa GTPase regulatory factor binding protein 1,RPGRIP1L;也称为肾单位肾痨(nephronophthisis,NPHP)]或Coiled-Coil and C2 Domain Containing 2A(CC2D2A)等基因的突变可导致COACH综合征,其中TMEM67为最主要致病突变,是与眼部结构缺损最相关的基因[15,19-22]。已有研究表明TMEM67基因是通过非经典的Wnt信号传导引起细胞过度增殖,凋亡增加,小脑、视网膜等发育不全和萎缩,导致一系列相关临床表现[1,23-24]。本研究中COACH综合征患儿眼部病变尤为突出,出现了眼球不自主震颤、视网膜色素异常及结构缺失等。既往研究表明眼部病变除了与TMEM67突变密切相关外,还与Abelson辅助质体整合位点1(abelson helper integration site-1,AHI1)、肌醇多聚磷酸5-磷酸酶(inositol polyphosphate-5-phosphatase,INPP5E)、ADP核糖基化因子样GTPase 13B(ADP-ribosylation factor-like GTPase 13B,ARL13B)、跨膜蛋白237(transmembrane protein 237,TMEM237)、Talpid3基因及CC2D2A相关,其中AHI1基因突变与眼球震颤最密切相关[15,25]。
对本研究的先证者及其家系全外显子进行分析,检出TMEM67基因c.725A>G(p.N242S)和c.1288+1G>A(splicing)复合杂合突变,其中c.725A>G(p.N242S)突变的致病性已在Romani 等[25]研究中得以验证,但c.1288+1G>A(splicing)突变目前未见报道;根据美国医学遗传学和基因组学院(The American College of Medical Genetics and Genomics, ACMG)指南[26],c.725A>G(p.N242S)变异评级为疑似致病性变异(likely pathogenic),c.1288+1G>A(splicing)突变评级为疑似致病性变异,且遗传学特征符合孟德尔遗传定律[27],结合患儿典型临床表现,符合该病特征,在全外显子基因分析中没有发现与发育延迟相关的其他潜在致病变异,表明该新突变位点是导致COACH综合征的新突变位点。
目前国内外文献中COACH综合征部分成年患者是出现消化道出血就诊才被诊断为COACH综合征,在治疗上还处于对症治疗阶段,例如内镜止血、护肝药物口服等治疗[19,25],因此早期的诊断对改变患者的预后有一定意义。
本研究中COACH综合征患儿检出c.1288+1G>A(splicing) ,扩展了TMEM67的突变谱;通过对患儿准确的诊断及其家系基因分析,可帮助家属进行遗传咨询,通过遗传疾病的早期管理能够延缓疾病的进程,从而为产前诊断提供依据。同时通过本病例报告补充了该病的基因库,对提高临床医师诊疗提供了一定的帮助。
