临床试验中的成组序贯设计
2022-12-23胥芹张怡君田雪夏雪张晓丽李静王安心
胥芹,张怡君,3,田雪,3,夏雪,张晓丽,李静,王安心
1 背景
在临床试验中,传统的试验设计方法如平行组设计、交叉设计和析因设计均属于固定设计,即需在完成所有受试者的主要结局随访后再对试验数据进行分析和评价。从临床试验的伦理和成本效益角度来看,研究者们通常希望在试验过程中进行期中分析,以比较处理组间的疗效和安全性,并评估各中心的试验状况、试验数据质量等。成组序贯设计能够早期终止试验,缩短试验时间,减少试验所需样本量,加速有效药的批准和应用,是期中分析中应用较为广泛的方法。
传统序贯设计方法是将每一对新的受试者随机分配接受不同的干预,在完成试验后进行一次统计分析,根据结果决定是否终止或继续试验。但是该方法的可操作性较差。Pocock[1]于1977年在传统序贯设计的基础上提出了成组序贯方法,随后O’Brien-Fleming和Lan等学者对该方法进行了改进[2-3]。与传统临床试验相比,成组序贯设计将多次假设检验的思想纳入到试验设计中来,样本量估算、统计分析等方面不同于既往传统临床试验。本文以脑血管病临床试验相关实例为基础,对成组序贯设计的设计思路、与传统临床试验的比较、使用场景、案例解读、设计要点和报告规范等方面进行简单的介绍。
2 设计思路
成组序贯设计是每间隔一定的时间或一定比例的样本量后对已完成试验的所有受试者进行期中分析,以判断试验是否可以提前得到有效或无效结论。从设计方案角度来看,成组序贯设计是将整个试验划分成K个连续的时间段,每个时间段内的每个处理组都有n个受试者加入。当第K个阶段(K=1,2,……,K)结束后,把之前所有阶段试验结果累积起来进行一次统计分析。对于拒绝性的成组序贯设计,如果拒绝H0则试验结束,否则继续下一阶段试验。当其处于最后一个试验阶段时,其结果不是接受H0,就是拒绝H0[4]。
成组序贯设计的一般步骤:
(1)根据试验的数据类型(计量、计数或生存资料等)选择合适的统计分析方法,指定相应的统计模型和效应参数θ,提出原假设:H0∶θ=0;指定试验的总Ⅰ类错误、容许误差和检验效能。
(2)选择成组序贯设计的方法:如固定界值法中的Pocock法、O’Brien-Fleming法或损耗函数法等,并确定试验的总阶段数。
(3)在满足总Ⅰ类错误和检验效能的概率条件下,计算对应的成组序贯设计每阶段的边界值参数和试验所需的最大信息量,并进一步得到所需的样本量。
(4)收集每阶段的数据并进行统计分析,计算当前阶段的检验统计量并进行假设检验,若拒绝原假设则停止试验并给出结论,否则继续进行下一阶段试验;在试验的最后阶段,若不能得出拒绝原假设的结论,则接受原假设并终止整个试验。
3 成组序贯设计与传统临床试验的比较
采用成组序贯设计的临床试验与传统临床试验在研究目的、研究设计、盲法、样本量计算、试验方案、统计分析计划等方面均有不同程度的差异,两者的区别见表1。
4 使用场景
根据国家药品监督管理局公布的药物临床试验的生物统计学指导原则[5],成组序贯设计一般用于创新药物的临床试验,而不用于仿制药的临床试验。当怀疑试验药物有较高的不良反应发生率,或研究者和申办方对试验药物预期疗效或预期疗效差均有很大的不确定性,为了避免试验失败造成的大量资金浪费,可考虑采用在期中分析中基于有效性或无效性的结果提前结束试验。当试验药与对照药的疗效相差较大,但病例稀少或临床观察时间过长,采用成组序贯设计可以根据其有效性提前终止试验。
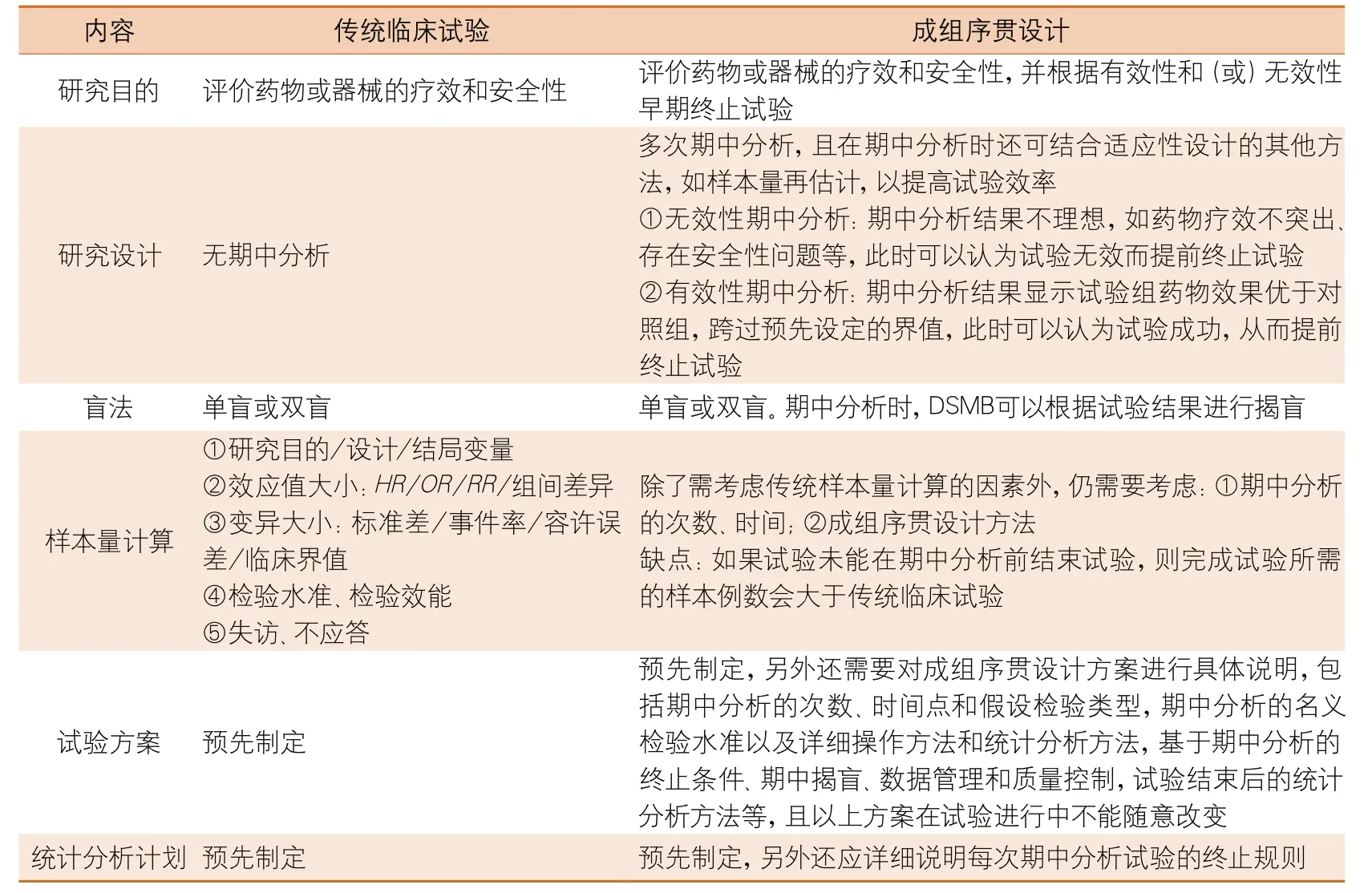
表1 成组序贯设计与传统临床试验的比较
5 案例解读
5.1 案例1 急性脑出血的降压治疗试验(antihypertensive treatment of acute cerebral hemorrhage 2,ATACH-2)研究是一项国际多中心、随机、开放标签的Ⅲ期临床试验。该试验在发病4.5 h内的自发性幕上脑出血患者中比较强化降压治疗是否优于标准降压治疗。该研究纳入年龄≥18岁、发病时间≤4.5 h、血肿<60 mL且GCS评分≥5分、发病后至少有一次收缩压≥180 mmHg(1 mmHg=0.133 kPa)的自发性幕上脑出血患者,研究对象被随机分配至强化强压治疗组或标准降压治疗组,使用静脉注射尼卡地平进行降压,在随机化后的24 h内分别将2组的每小时的最低收缩压控制在110~139 mmHg和140~179 mmHg,主要终点指标为3个月时中重度残疾或死亡情况(mRS 4~6分)[6]。
ATACH-2研究采用成组序贯设计方法,分别在收集完1/3和2/3样本量的主要终点指标随访时进行2次基于有效性和无效性的期中分析。根据既往文献报道,研究者估计标准降压治疗组脑出血后3个月死亡或残疾发生率为60%,强化降压治疗组为50%,RR为0.83,试验组与对照组样本量比例为1∶1,总Ⅰ类错误和检验效能分别取0.05和90%,预计需要样本量1042例。考虑失访率或治疗失败引起的不依从率(R=10%),根据公式1/(1-R)2得到膨胀因子1.23,最终研究所需的样本量为1280例,每组各640例。
在第1次期中分析后,数据与安全监察委员会(Data and Safety Monitoring Board,DSMB)建议在1/2样本量时增加一次计划外的期中分析。研究者采用O’Brien and Flemingα消耗函数法的边界值作为有效性判定条件,4次分析的名义检验水准分别为0.0002、0.0030、0.012和0.046。采用基于条件检验效能(conditional power,CP)的随机缩减法作为试验无效的判定标准,当CP低于某一值(如20%)后,DSMB将会对整个试验进行评估,包括入组率、次要结局指标数据等,从而判定试验是否能够由于无效而早期终止。在第1次分析后,DSMB决定在第2次和第3次期中分析时将CP低于20%作为试验无效性的判定边界。当试验完成850例(2/3样本量)患者的90 d随访时,在没有揭盲的情况下,研究者分别计算了基于2组作为参照组的CP,分别为9.11%和4.73%且均低于预设的20%,所以DSMB决定停止试验。
5.2 案例2 直接血管内血栓切除术与静脉溶栓(intravenous thrombolysis,IVT)联合血管内血栓切除术治疗前循环急性大血管闭塞患者的疗效(direct endovascular thrombectomyvs.combined IVT and endovascular thrombectomy for patients with acute large vessel occlusion in the anterior circulation,DEVT)这一研究是在急性颅内大血管闭塞卒中患者中比较单独血管内治疗与阿替普酶静脉溶栓桥接血管内治疗有效性及安全性的研究,是一项多中心、随机、开放标签、结局盲法的非劣效临床试验,旨在判断对发病4.5h内大血管闭塞所致急性缺血性卒中患者进行单独血管内取栓是否不劣于桥接取栓,研究的主要结局指标是90 d良好功能预后(mRS 0~2分)[7]。
DEVT试验采用成组序贯设计方法,计划在完成20%、40%、60%、80%和100%样本量时进行期中分析。根据既往文献报道,研究者估计桥接取栓组90 d良好功能预后为43%,非劣效界值设定为-10.0%,2组样本量比例为1∶1,单侧α=0.025,β=0.2,考虑5%的失访率,最终研究所需的样本量为970例。该研究采用Pocockα消耗函数法来计算样本量和调整Ⅰ类错误,5次分析的名义检验水准分别为0.009 18、0.007 10、0.009 31、0.007 84和0.009 05。在完成20%的样本量即194例时,2组的检验P=0.008小于预先设定的界值(P=0.0918),非劣效成立,DSMB决定试验提前终止。
6 设计要点
6.1 期中分析的次数 期中分析次数的确定在成组序贯试验中需要慎重考虑,多次的期中分析会增大试验总样本量,耗费大量人力、财力。更重要的是,为了控制试验的总Ⅰ类错误,多次的期中分析需要校正分配到各期的名义检验水准,使其值变得更小,除非试验药物效果非常优越,显著性检验结果有很大差异的预期,否则试验在前期将会更难得到中止。一般期中分析次数以不超过5次为宜。
6.2 期中分析的时间 时间点的选择有日历时间和信息时间2种方式,通常采用信息时间,即在累积完成某一比例的样本例数或事件数时进行分析。例如三阶段成组序贯试验预计死亡600例,可在观察到死亡人数300例、450例和600例时,即信息时间为0.5、0.75和1的时候进行统计分析。时间点选择的考量要求主要有:数据量是否充分,随访时间是否足够,是否有足够的疗效估计和安全性评价结果,也要考虑重要的次要结局和重要的亚组信息是否足够。
6.3 Ⅰ类错误的调整方法 在成组序贯设计试验中,若多次假设检验都是采用0.05的双侧显著性检验水准,则会导致Ⅰ类错误膨胀,假阳性的可能性增加。因此需要对各阶段的检验水准进行校正,校正后的检验水准称为名义检验水准。
目前常用的校正方法包括固定界值法和α消耗函数类。固定界值法包括Pocock法、O’Brien-Fleming法和Peto法。这三种方法的比较见表2。
由于Pocock法和O’Brien-Fleming法要求必须先确定期中分析的次数,并且每次期中分析增加的患者数应该大致相似。Lan等[3]于1983年提出了一种更为灵活的α消耗函数方法。常用的损耗函数形式有以下三种。其中①所计算的期中分析界值和名义检验水准近似于Pocock设计公式,②则近似于O’Brien-Fleming设计,因此,它们也被成为Pocockα消耗函数和O’Brien-Flemingα消耗函数,并在实际临床试验应用和统计软件中替代Pocock设计和O’Brien-Fleming设计使用。
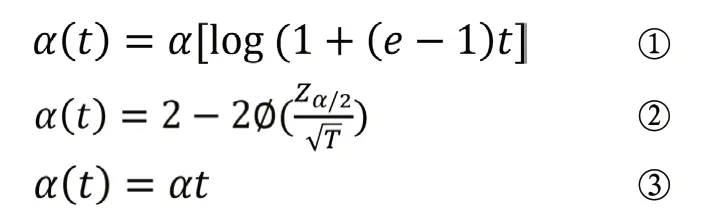
6.4 试验终止规则 目前成组序贯设计常采用的方法是α消耗函数法,也有文献报道了随机缩减法,其中以CP作为衡量指标最为常用。CP是指通过当前阶段所纳入样本提供的信息,估计试验完成时能够得出有效结论的可能性[8]。在研究开始之前,需要提前设定有效性界值和无效性界值。有效性界值是将Ⅰ类错误率维持在指定水平之下,通常使用α消耗函数计算,越过边界意味着已经越过统计显着性阈值,试验有效。无效性界值是使检验效能保持在指定水平之上,通常使用β消耗函数或CP计算,越过边界意味着检测到具有统计学意义的结果的可能性已经低于期望值,试验无效。
7 报告规范
成组序贯设计属于适应性设计的一种,报告规范可参考《试验报告统一标准》针对适应性设计的扩展版——适应性设计试验报告统一标准拓展[9]。
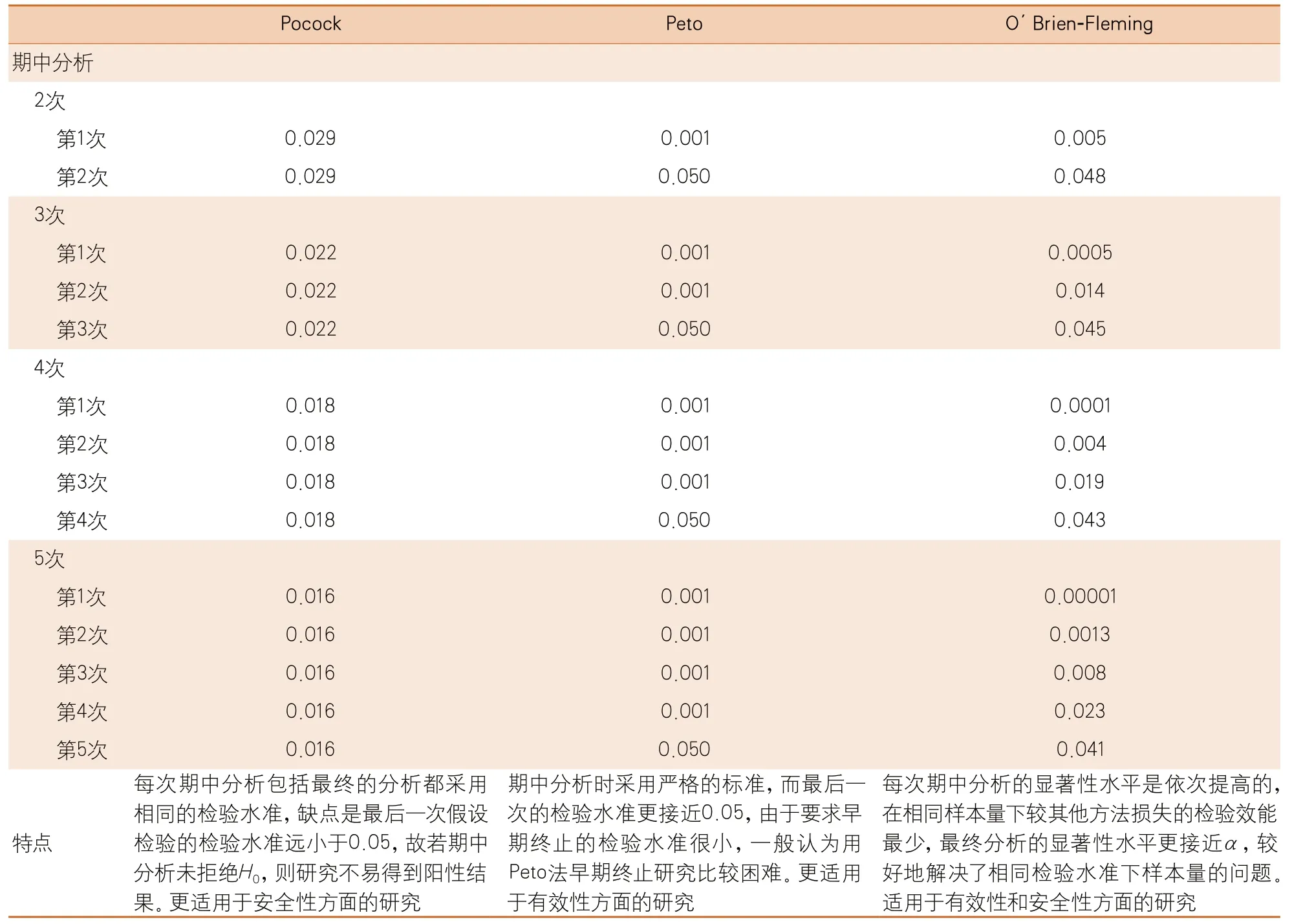
表2 常用的固定界值法的比较(总α=0.05)
