人工设计短肽PF1和PF2的克隆表达及其对葡萄糖氧化酶活性的影响
2022-12-22李传博鲁明杰张庆芳林禹彤窦少华
李传博,鲁明杰,张庆芳,林禹彤,窦少华
(大连大学生命健康学院,辽宁省海洋微生物工程技术研究中心,辽宁 大连 116622)
葡萄糖氧化酶(glucose oxidase,GOD)是一种氧化还原酶[1],以分子氧为电子受体,与过氧化氢酶形成氧化还原系统,专一性催化β-D-葡萄糖转化为葡萄糖酸[2-4]。GOD的催化效率和反应速率通常受各种因素影响,如酸度和碱度、温度、氧气和动力学参数[5-7]。GOD作为一种高特异性氧化还原酶,在食品领域发挥着极其重要的作用[8-11]。例如GOD常用于保鲜剂。马清河等[12]发现,GOD对鲜虾具有抗褐变保鲜作用,冷藏(-18 ℃)12个月后仍能保持二级鲜度。GOD作为保鲜剂抑制鲜虾褐变的现象为是否可以通过设计不同带电短肽影响酶的催化作用提供了新的思路。
早在2000年,李菲等[13]便基于表面静电互补的方法建立短肽抑制剂与酶识别的模式。陈瑞华[14]发现胰蛋白酶、蛋白酶K和α-糜蛋白酶水解杆菌肽底物时,Zeta电位绝对值增加,并且酶抑制剂的添加增加了胰蛋白酶和蛋白酶K的Zeta电位值,负电荷的增加会抑制酶与底物的酶促反应。由此可知,添加具有不同电荷性质蛋白质可以直接影响酶促反应进程。刘荣娜[15]研究了不同带电蛋白质对酶促反应的影响表明,酶活力的改变与不同带电蛋白质浓度呈正相关,并提出加入与酶带同种电荷的蛋白质,并且当蛋白质与酶形成一定距离时会对酶本身产生微弱的斥力,使酶在酶促反应中更好地发挥催化活性。本研究将PF1、PF2两条短肽的基因在大肠杆菌(Escherichia coli)BL21中克隆表达,产物经Ni-NTA纯化后加入GOD酶促反应,并对GOD活力及Zeta电位进行检测,旨在为今后研究肽链PF1、PF2对GOD之间相互作用提供物质基础,可以为酶的食品、医药、农业应用等研究提供新的科学依据。
1 材料与方法
1.1 材料与试剂
E. coliBL21(DE3)用于pET系列载体的诱导表达,质粒pET-30a(+)为异丙基-β-D-硫代半乳糖苷(isopropyl-β-D-thiogalactopyranoside,IPTG)诱导的表达载体,均保藏于辽宁省海洋微生物工程技术研究中心。
Pyrobest DNA Polymerse、NdeI、Hind III限制性内切酶、T4 DNA ligase、Kan、TaKaRa MiniBEST Plasmid Purification试剂盒 TaKaRa(大连)有限公司;BM5000 DNA ladder 北京鼎国昌盛生物技术有限责任公司;DNA Marker DL2000 北京全式金生物技术有限公司;胰蛋白胨、酵母提取物 生工生物工程(上海)股份有限公司;Ni-NTA His·Bind Resins 中国科学院过程工程研究所;蛋白定量试剂盒 美国Bio-Rad公司;正己烷(色谱纯) 德国Meker公司;GF254薄层色谱硅胶 青岛海洋化工厂;所有分离用有机溶剂均为国产分析纯。
培养基及溶液:LB(Luria-Bertani)培养基、STE缓冲液、TBST(Tris-HCl+Tween)缓冲液、PBST(PBS+Tween)缓冲液、TE(Tris-EDTA)缓冲液、TAE(Trisacetate-EDTA)缓冲液等均参考《精编分子生物学实验指南》[16]和《分子克隆实验指南》[17]附录部分以及相关文献[18]配制。
1.2 仪器与设备
MilliQ plus超纯水系统 美国Millipore公司;CR-22E高速冷冻离心机 日本Hitachi公司;LTI-700低温恒温培养箱 上海爱朗仪器有限公司;JY99-IIIBN超声波连续流细胞粉碎机 宁波新芝生物科技股份有限公司;PowerPac 300蛋白电泳仪 美国Bio-Rad公司;Multiskan GO全波长酶标仪 美国Thermo公司;FE20/EL20 pH计 瑞士Mettler Toledo公司。
1.3 方法
1.3.1 人工设计肽链PF1、PF2基因序列的获得
根据E. coliBL21(DE3)的密码子偏好性[19],人工设计并合成了2 条带有不同电荷肽链基因PF1、PF2,等电点分别为12.01、3.18,基因全长均为309 bp。
1.3.2 引物的设计与合成
采用primer 5.0软件对PF1、PF2序列进行比对分析后,对照pET-30a(+)载体上的多克隆酶切位点,设计基因的特异性聚合酶链式反应(polymerase chain reaction,PCR)扩增引物PF1-F、PF1-R和PF2-F、PF2-R[20],见表1。引物由北京鼎国昌盛生物技术有限责任公司合成。

表1 本研究所用引物Table 1 Primers used in this study
1.3.3 构建重组表达载体
PCR体系50 μL,用正向引物PF1-F、PF2-F和反向引物PF1-R、PF2-R进行扩增[21]。通过1 g/100 mL琼脂糖凝胶电泳检测并回收目的条带。PF1、PF2通过T4 DNA于4 ℃连接反应过夜至pET-30a(+)载体。然后转化至感受态细胞E. coliBL21(DE3),用TaKaRa Mini BEST Plasmid Purification试剂盒提取阳性克隆。PCR扩增该基因,PCR产物通过NdeI和HindIII双酶切鉴定。
1.3.4 人工设计短肽PF1、PF2在E. coliBL21(DE3)的诱导表达
将测序正确的重组菌命名为pET-30a(+)-PF1/BL21、pET-30a(+)-PF2/BL21。转化子接入5 mL LB培养基中,37 ℃、200 r/min条件下过夜培养。次日,取1 mL过夜菌接种至100 mL新LB培养基中,37 ℃、200 r/min培养至菌液OD600nm为0.6~0.8左右,加入终浓度为0.1 mmol/L的IPTG,pET-30a(+)-PF1/BL21与pET-30a(+)-PF2/BL21分别以诱导15 ℃、160 r/min培养16 h和37 ℃、160 r/min培养4 h[22]。离心收集菌体,并用Binding Buffer洗涤3次。加入5 mL Binding Buffer重悬细胞,超声波破碎后(开5 s、关4 s,时间40 min),4 ℃、12 000 r/min离心30 min,舍掉沉淀获得粗蛋白溶液[23]。
1.3.5 Ni-NTA纯化粗蛋白溶液
由pET-30a(+)载体表达的靶蛋白具有6个组氨酸的蛋白质标签,组氨酸可以与Ni2+鳌合,从而使带有组氨酸标签的靶蛋白质与Ni-NTA纯化介质结合。诱导表达后的粗酶液上样Ni-NTA柱,并用40 mmol/L咪唑缓冲液洗柱子,直到考马斯亮蓝蓝色不变为止。取上述步骤中收集的100 μL溶液,加入100 μL 2×蛋白电泳上样缓冲液;沸水煮10 min,14 000 r/min离心5 min,取上清液进行十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDS-PAGE)检测[24-26]。
1.3.6 纯化后蛋白PF1和PF2 Zeta电位检测
开启Zeta电位仪,设置仪器温度为35 ℃,预热30 min。将1.4 mL 0.004 mg/mL的GOD溶液加入到Zeta电位样品池中,测定Zeta电位。分别将1.4 mL 1 mg/mL的PF1和PF2溶液加入到Zeta电位样品池中,测定Zeta电位。分别将0.7 mL 2 mg/mL的PF1和PF2溶液加入到0.7 mL 0.008 mg/mL的GOD溶液中处理5 min,测定GOD Zeta电位。
1.3.7 纯化后蛋白PF1和PF2对GOD活力影响
分别将1 mg/mL的肽链PF1和PF2溶液加入到0.1 mL 0.004 mg/mL的GOD溶液中处理5 min,测定酶活力,将0.1 mL磷酸缓冲液同样加入到0.1 mL 0.004 mg/mL的GOD溶液中处理5 min作为对照,测定酶活力。在实验温度和pH值下,60 s内将1.0 μmol催化葡萄糖氧化成为H2O2和葡萄糖酸所需要的GOD量定义为一个酶活力单位[3]。
GOD活力测定:酶标仪设定为500 nm波长处连续进行300次吸光度测定,每秒测定一次。分别在试管中加入2.5 mL邻联茴香胺缓冲液、0.3 mL葡萄糖溶液和0.1 mL过氧化氢酶溶液并混匀,放置水浴锅中给定温度水浴5 min。将0.1 mL GOD溶液和0.1 mL磷酸缓冲液迅速加入混合溶液中,在20 s内分别将3个0.31 mL混合液加入到96 孔板中测定吸光度,根据下式计算GOD活力:
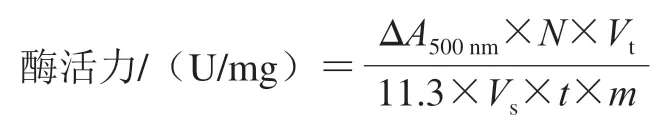
式中:ΔA500nm为每秒吸光度的降低值;N为酶液的稀释倍数;Vt为样品反应的总体积/mL;11.3为消光系数(L/mol);Vs为稀释后酶液加入体积/mL;t为反应时间/min;m为GOD样品质量/g。
2 结果与分析
2.1 人工设计肽链PF1、PF2基因的PCR扩增
利用特异性引物PF1-F、PF1-R和PF2-F、PF2-R,使用PF1、PF2基因DNA作为模板扩增目的片段,大小约为309 bp(图1),与预计的片段大小相近。切胶回收并测序,确定为目的基因的DNA片段。

图1 PF1(A)、PF2(B)基因片段的电泳检测Fig. 1 Argarose gel electrophoresis of the conserved region fragments of PF1 (A) and PF2 (B)
2.2 重组质粒pET-30a(+)-PF1、pET-30a(+)-PF2的构建及鉴定
2.2.1PF1、PF2基因PCR产物双酶切
用NdeI/Hind III双酶切PF1、PF2基因PCR产物,取1 μL产物进行1%琼脂糖凝胶电泳,图2结果显示,在750 bp附近出现特异性条带与理论设计值相符。

图2 PF1和PF2基因PCR产物双酶切的电泳检测Fig. 2 Detection of PF1 and PF2 PCR products by double enzyme digestion
2.2.2 原核表达质粒pET-30a(+)双酶切
用NdeI/Hind III双酶切原核表达载体pET-30a(+),取1 μL进行1%琼脂糖凝胶电泳,结果如图3所示。质粒pET-30a(+)双酶切产物条带单一,且在5 000~6 000 bp之间,符合预期。
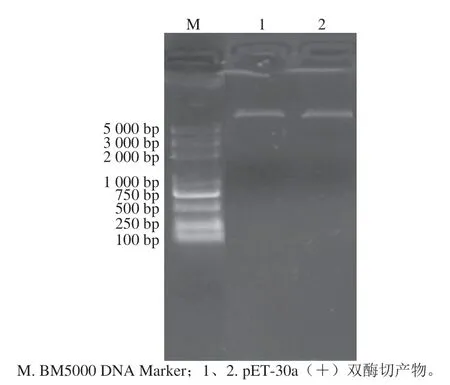
图3 原核表达质粒pET-30a(+)双酶切Fig. 3 Identification of prokaryotic expression plasmid pET-30a(+) by double enzyme digestion
2.2.3 菌落PCR筛选阳性克隆
从pET-30a(+)-PF1/BL21、pET-30a(+)-PF2/BL21转化的平板上分别随机挑取17个白斑直接作为模板进行PCR扩增(图4),pET-30a(+)-PF1/BL21经电泳分析发现8号、9号和12号PCR产物与目的基因(309 bp)大小一致,表明8号、9号和12号成功将靶基因与表达质粒pET-30a(+)相连。pET-30a(+)-PF2/BL21经电泳分析发现4号、9号和15号PCR产物与目的基因(309 bp)大小一致,表明4号、9号和15号成功将靶基因成功连接到表达质粒pET-30a(+)上。
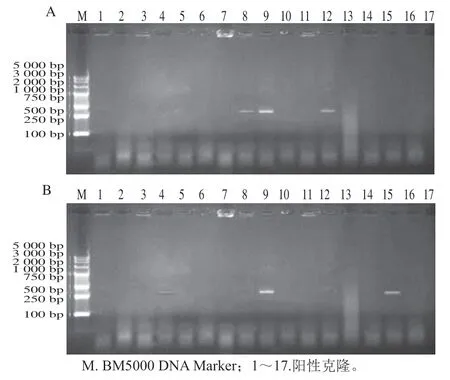
图4 菌落PCR验证PF1(A)、PF2(B)基因的电泳检测Fig. 4 Agarose gel electrophoresis analysis of positive clones of PF1 (A) and PF2 (B) selected by colony PCR
2.2.4 重组质粒的酶切鉴定和测序鉴定
通过质粒提取试剂盒提取pET-30a(+)-PF1、pET-30a(+)-PF2重组质粒,用限制性核酸内切酶NdeI/Hind III进行双酶切,酶解产物进行1%琼脂糖凝胶电泳检测,结果如图5所示。可以看出,重组质粒pET-30a(+)-PF1、pET-30a(+)-PF2酶切产物片段大小分别约为309 bp和5 422 bp,即表达的pET-30a(+)-PF1、pET-30a(+)-PF2和原核表达载体pET-30a(+)。说明PF1、PF2基因已成功亚克隆至表达载体pET-30a(+)中。

图5 pET-30a(+)-PF1(A)、pET-30a(+)-PF2(B)经Nde I/Hind III酶切产物Fig. 5 Agarose gel electrophoresis analysis of pET-30a(+)-PF1 (A) and pET-30a(+)-PF2 (B) digested with Nde I/Hind III
2.3 pET-30a(+)-PF1、pET-30a(+)-PF2重组质粒在E. coli BL21(DE3)中的表达
如图6所示,泳道4、5、7、8、10、11约11 kDa蛋白带与PF1的理论分子质量(11.1 kDa)一致,表明IPTG成功诱导蛋白表达。肽链PF1在15 ℃、0.1 mmol/L IPTG诱导16 h表达较好。与泳道3、6、9未诱导的阴性对照相比,重组菌pET-30a(+)-PF1/BL21在约11 kDa处表现出特异性蛋白带,重组蛋白成功表达,且可溶性较好。
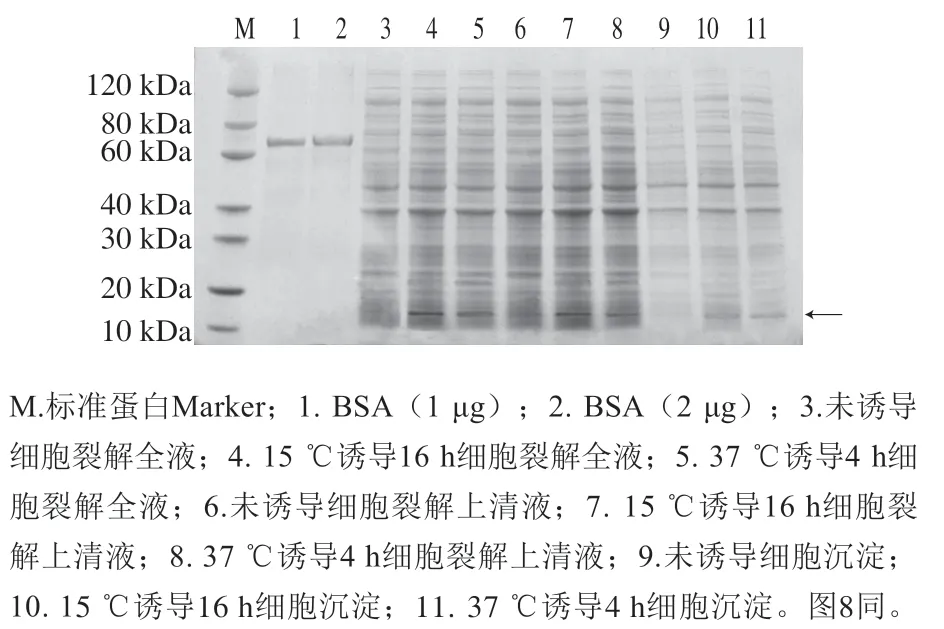
图6 重组pET-30a(+)-PF1的诱导表达SDS-PAGEFig. 6 SDS-PAGE analysis of PF1 expressed in BL21(DE3)
以His抗体为一抗,Western Blot分析鉴定表达产物免疫反应性。由图7可以看出,诱导产物在相同位置具有单个特异性条带。PF1蛋白分子大小与SDS-PAGE鉴定大小一致。
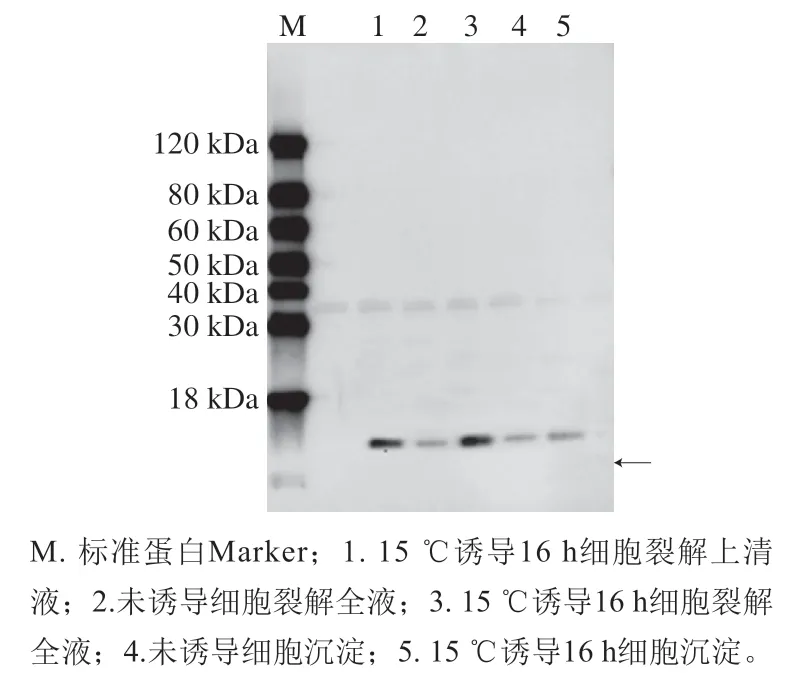
图7 重组质粒pET-30a(+)-PF1的Western Blot鉴定Fig. 7 Western blot analysis of PF1 expressed in BL21(DE3)
如图8所示,泳道4、5、7、8约11 kDa蛋白带与PF2的理论分子质量(11.1 kDa)一致,表明IPTG成功诱导表达蛋白表达。肽链PF2在37 ℃、0.1 mmol/L IPTG诱导4 h表达较好。与泳道3、6、9未诱导的阴性对照相比重组菌pET-30a(+)-PF2/BL21在约11 kDa处表现出特异性蛋白带,重组蛋白成功表达,且可溶性较好。

图8 重组pET-30a(+)-PF2的诱导表达Fig. 8 SDS-PAGE analysis of PF2 expressed in BL21(DE3)
以His抗体为一抗,Western Blot分析鉴定表达产物免疫反应性。由图9可以看出,诱导产物在相同位置具有单个特异性条带。PF2蛋白分子大小与SDS-PAGE鉴定大小一致。

图9 重组质粒pET-30a(+)-PF2的Western Blot鉴定Fig. 9 Western blot analysis of PF2 expressed in BL21(DE3)
2.4 pET-30a(+)-PF1、pET-30a(+)-PF2重组质粒Ni-NTA纯化
通过Ni-NTA亲和层析柱纯化PF1、PF2,SDS-PAGE检测洗脱的收集液,如图10所示。目标蛋白主要被300 mmol/L咪唑溶液洗脱下来,且洗脱液中无杂蛋白。

图10 重组pET-30a(+)-PF1与重组pET-30a(+)-PF2的纯化Fig. 10 SDS-PAGE analysis of purifeid recombinant pET-30a(+)-PF1 and pET-30a(+)-PF2
2.5 纯化后蛋白PF1和PF2对GOD Zeta电位检测
由表2可知,纯化后蛋白PF1表面Zeta电位为-18.16 mV,带负电荷,而PF2表面Zeta电位为10.32 mV,带正电荷。且经过PF1处理过的GOD电位由-14.60 mV下降至-16.07 mV,PF2处理过得GOD电位由-14.60 mV上升至-9.26 mV。

表2 纯化后蛋白PF1和PF2对GOD Zeta电位检测Table 2 Effect of purified proteins PF1 and PF2 on GOD zeta potential
2.6 纯化后蛋白PF1和PF2对GOD活力影响
由表3可知,使用纯化后带负电荷蛋白PF1处理后GOD活力提高了8.34%,带正电荷蛋白PF2处理后GOD活力降低了6.88%。证明本研究生物方法合成的带不同电荷蛋白对GOD活力同样有激活和抑制作用。

表3 纯化后蛋白PF1和PF2对GOD活力影响Table 3 Effect of purified proteins PF1 and PF2 on the activity of GOD
3 结 论
人工设计并合成了2 条肽链PF1、PF2,基因全长309 bp,以ATG作为起始密码子开始,以TGA作为终止密码子结束,编码93个氨基酸肽链。将其扩增至pET-30a(+)载体中,构建了重组质粒pET-30a(+)-PF1和pET-30a(+)-PF2。随后将酶切鉴定正确的表达质粒pET-30a(+)-PF1和pET-30a(+)-PF2转化至宿主细胞E. coliBL21(DE3)中,0.1 mmol/L IPTG大量诱导表达收集菌体细胞。通过超声破碎菌体细胞以获得粗酶溶液,利用重组质粒pET-30a(+)-PF1和pET-30a(+)-PF2上C端融合6个组氨酸的蛋白质标签,经Ni-NTA纯化分离纯化得到纯蛋白。SDS-PAGE与Western Blot分析表明:表达出的靶蛋白分子质量为11.1 kDa,这与预估大小一致。PF1和PF2人工设计蛋白的pI值分别为12.01、3.18,且表达产物以可溶性蛋白存在于细胞浆中。以上结果都指向同一结论:人工设计肽链基因PF1、PF2在E. coliBL21(DE3)中得到成功表达。
纯化后的蛋白PF1和PF2在pH 5.5的缓冲液中测定Zeta电位后发现,PF1 Zeta电位为-18.16 mV,带负电;PF2 Zeta电位为10.32 mV,带正电。通过使用纯化后的蛋白PF1和PF2处理GOD后,GOD活力出现了明显的变化,今后可以设计并合成更多的肽链对GOD进行处理,检测其对GOD活力的影响,并检测肽链与GOD的相互作用。短肽PF1和PF2的基因克隆及表达为后续将短肽PF1和PF2应用于大规模食品或工业化生产新型酶激活剂打下坚实基础。
