基于高通量测序数据挖掘探讨小鼠射血分数保留心力衰竭的发病机制
2022-12-20尤红俊赵倩倩任淑婷刁佳宇苟棋玲董梦雅
尤红俊,赵倩倩,任淑婷,刁佳宇,苟棋玲,董梦雅
射血分数保留心力衰竭(heart failure with preserved ejection fraction,HFpEF)是一种除以高血压、心肌肥厚和舒张功能障碍为特征的血流动力学紊乱之外,伴肥胖症和糖尿病等参与的多系统综合征,涉及心、肺、肾、骨骼肌、脂肪组织、血管系统、免疫和炎症反应等。随着人均寿命延长,迄今为止其占心力衰竭总人群近2/3[1]。目前,心肌肥厚、心肌纤维化、兴奋性收缩偶联缺陷、肌节功能障碍、氧化-氮化应激、微血管功能不全、炎症、线粒体和代谢缺陷等多因素被认为可能参与HFpEF 病理生理学过程[2]。因相关的并存疾病引起的表型复杂,其病理生理机制仍不甚清楚,故也缺乏针对病因及机制的有效治疗措施,其死亡率仍偏高,2 年内全因死亡率约35%,造成严重的社会经济学问题[3]。因而,探讨其发病机制将有助于临床诊疗工作。
既往关于HFpEF 的基础研究多基于高血压、糖尿病或高龄等单一因素诱导的心脏舒张功能障碍;而高脂饮食联合血管紧张素Ⅱ双重因素诱导的HFpEF 动物模型,可更近似模拟人群HFpEF 的复合表型,如糖脂代谢紊乱、氧化/氮化应激等,且其机制探索性研究尚未见报道。本研究基于基因表达综合数据库(Gene Expression Omnibus,GEO)来源的HFpEF 模型小鼠左心室组织转录组高通量测序数据,应用生物信息学手段,寻找疾病差异表达基因(differentially expressed genes,DEGs),构建蛋白质-蛋白质相互作用(protein-protein interaction,PPI)网络,鉴定核心基因,再进行富集分析及疾病关联性分析,探讨HFpEF 发病机制,从而为疾病诊疗提供理论支持。
1 资料与方法
1.1 数据下载 在GEO 数据库(www.ncbi.nlm.nih.gov/GEO)[4-5]中搜索“HFpEF”,获取高通量转录谱测序数据集GSE153923,其来源于荷兰赫布雷希特研究所发育生物学和干细胞研究系,由Withaar 等[6]研究者提供。该数据集以高脂饮食联合血管紧张素Ⅱ干预雌性C57BL/6J 小鼠(18~22 月龄)构建HFpEF 模型,作为实验组(样本量为4),以月龄、体重匹配的雌性小鼠低脂饮食饲养作为对照组(样本量为4),取左心室组织标本进行转录组测序分析。采用GPL19057 Illumina NextSeq 500(Mus musculus)平台。
1.2 差异表达基因分析 采用统计软件R 语言(版本R 4.1.1)“DESeq2”包[7]进行基因差异表达分析。以|log fold change|≥1,adjustedP<0.05 为DEGs 的筛选条件,以R 语言“ggplot2”包(https://cran.r-project.org/web/packages/ggplot2/)绘制火山图以可视化DEGs。
1.3 PPI 网络分析 将DEGs 导入STRING(Search Tool for the Retrieval of Interacting Genes,http://string-db.org/cgi/input.pl)进行PPI 网络分析[8]。设定可信度(confidence)为0.4,输出TSV 格式文件,用于后续Cytoscape 分析筛选核心基因[9]。
1.4 核心基因鉴定 使用Cytoscape 3.7.3 软件插件CytoHubba (http://hub.iis.sinica.edu.tw/cytohubba/)筛选出PPI 网络中的核心基因[10]。拓扑评分策略之一Degree 代表基因/蛋白间相互作用关系连通度,以Degree值排序前30 位的基因作为PPI 网络中的核心基因。
1.5 基因本体(GO)和京都基因与基因组百科全书(KEGG)富集分析 使用R 语言对DEGs 和核心基因分别进行GO(http://www.geneontology.org)和KEGG通路富集分析[11-12]。GO 富集分析分别在生物学过程(biological processes,BP)、细胞组分(cellular component,CC)和分子功能(molecular function,MF)3 个方面对基因进行功能注释[13]。利用R 语言“ggplot2”包将结果可视化。P<0.05 被认为差异有统计学意义。
1.6 核心基因与心力衰竭(舒张性)的相互作用 利用比较毒理基因组学数据库(Comparative Toxicogenomics Database,CTD,http://ctdbase.org/)的数据分析核心基因与心力衰竭(舒张性)风险之间的关系。CTD 整合了包括化合物-基因/蛋白质相互作用、化合物-疾病和基因-疾病关系在内的信息,以探索与疾病发病机制相关的假设[14]。
2 结 果
2.1 基因差异表达分析 以Log(fold change)≥1,adjustedP<0.05 为标准,与对照组比较,HFpEF 小鼠左心室组织中337 个基因存在差异表达,其中,212个表达上调,125 个表达下调。基因差异表达谱的火山图见图1。
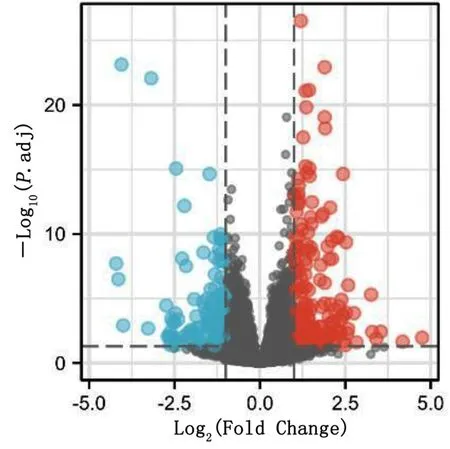
图1 差异基因表达谱的火山图
2.2 DEGs 富集分析 DEGs GO 富集分析提示,在BP 方面,DEGs 主要参与细胞外结构和基质的构成、损伤应答和愈合、胶原纤维构成、血液凝固和细胞趋化性等;在CC 方面,DEGs 主要参与细胞外基质、含胶原的细胞外基质、细胞外基质成分、胶原三聚体和带状胶原纤维等;在MF 方面,DEGs 主要参与细胞外基质结构成分、糖胺聚糖结合、受体配体活性、纤连蛋白结合和肝素结合等。详见图2。
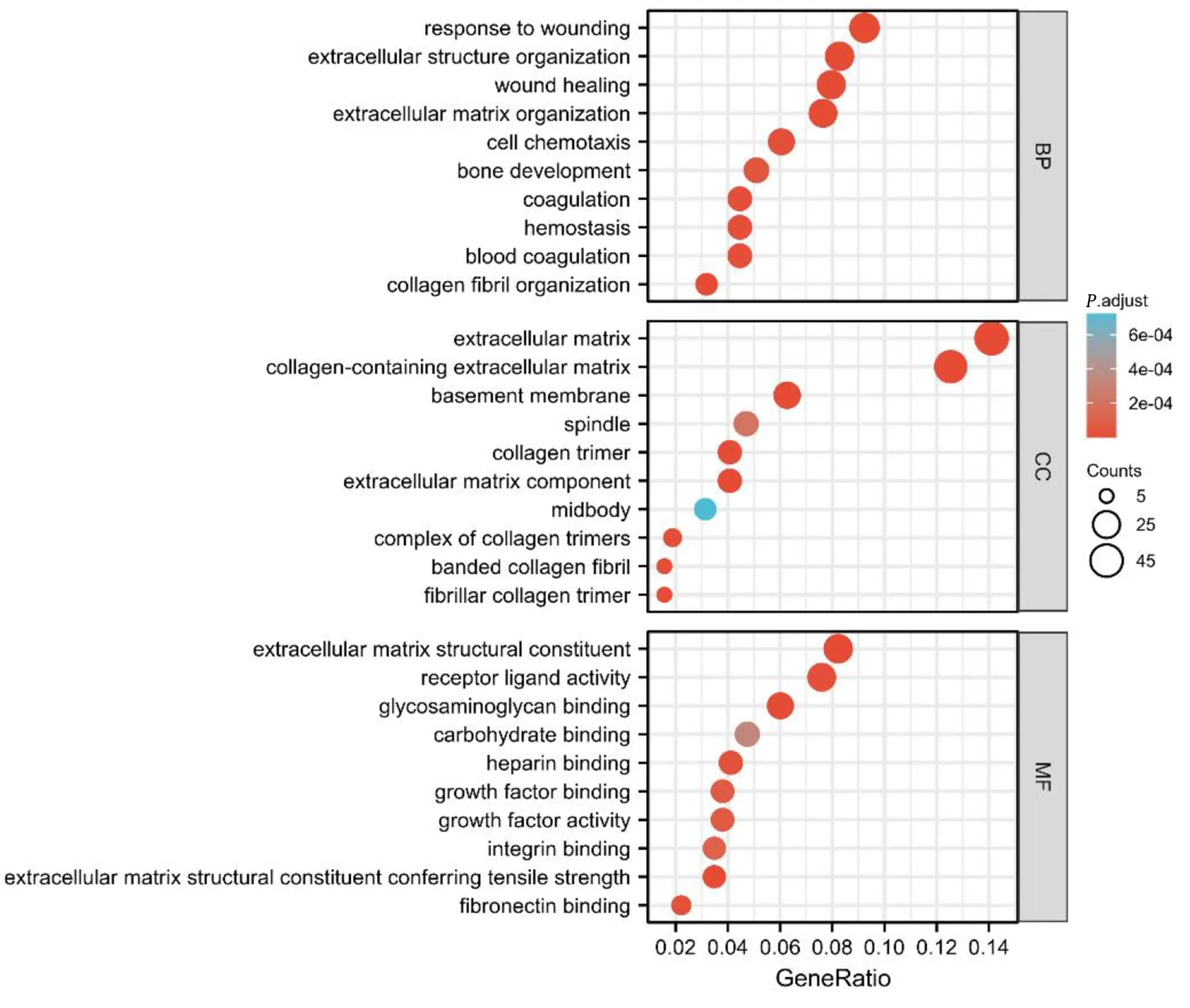
图2 DEGs GO 富集分析部分可视化(气泡图)
DEGs KEGG 富集分析提示,DEGs 参与细胞外基质-受体相互作用、黏着斑、磷脂酰肌醇3-激酶/蛋白激酶B(PI3K-Akt)、血小板活化、糖尿病并发症中的晚期糖化终产物(AGEs)及其受体(RAGE)、缺氧诱导因子(hypoxia-inducible factor,HIF)1 等信号通路中。详见图3。
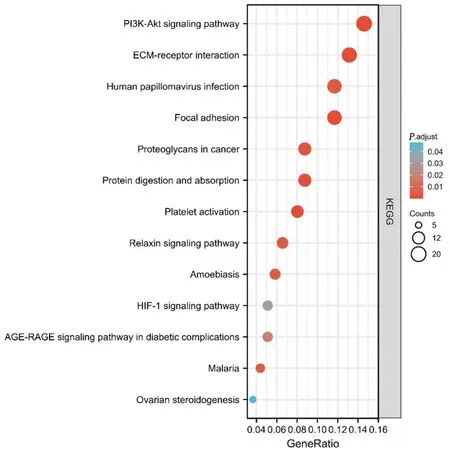
图3 DEGs KEGG 富集分析部分可视化(气泡图)
2.3 PPI 网络及核心基因鉴定 对337 个DEGs 采用STRING 在线数据库进行PPI 网络分析,构建PPI 网络,隐去离散节点后,所构建的PPI 网络中节点(蛋白数)321 个、边数(蛋白间作用关系)1 183 条,平均节点度值为7.37,PPI 富集P值<1.0e-16。输出TSV 格式文件并导入Cytoscape 软件,进行cytoHubba 运算,筛选出Degree 值排名前30 位及与其直接作用的节点,构成子网络,节点数为128 个,边数为998 条,详见图4。Degree 值排名前30 位的鉴定为核心基因,即Fn1、Mki67、Mmp9、Col1a1、Ctgf、Col1a2、Col3a1、Timp1、Cdk1、Hmmr、Foxm1、Racgap1、Ckap2、Spag5、Ncapg、Cenpe、Fbn1、Spp1、Thbs1、Shcbp1、Cdca3、Ckap2l、Tpx2、Cenpf、Kif20a、Cep55、Prc1、Ect2、Lox 和Anln。
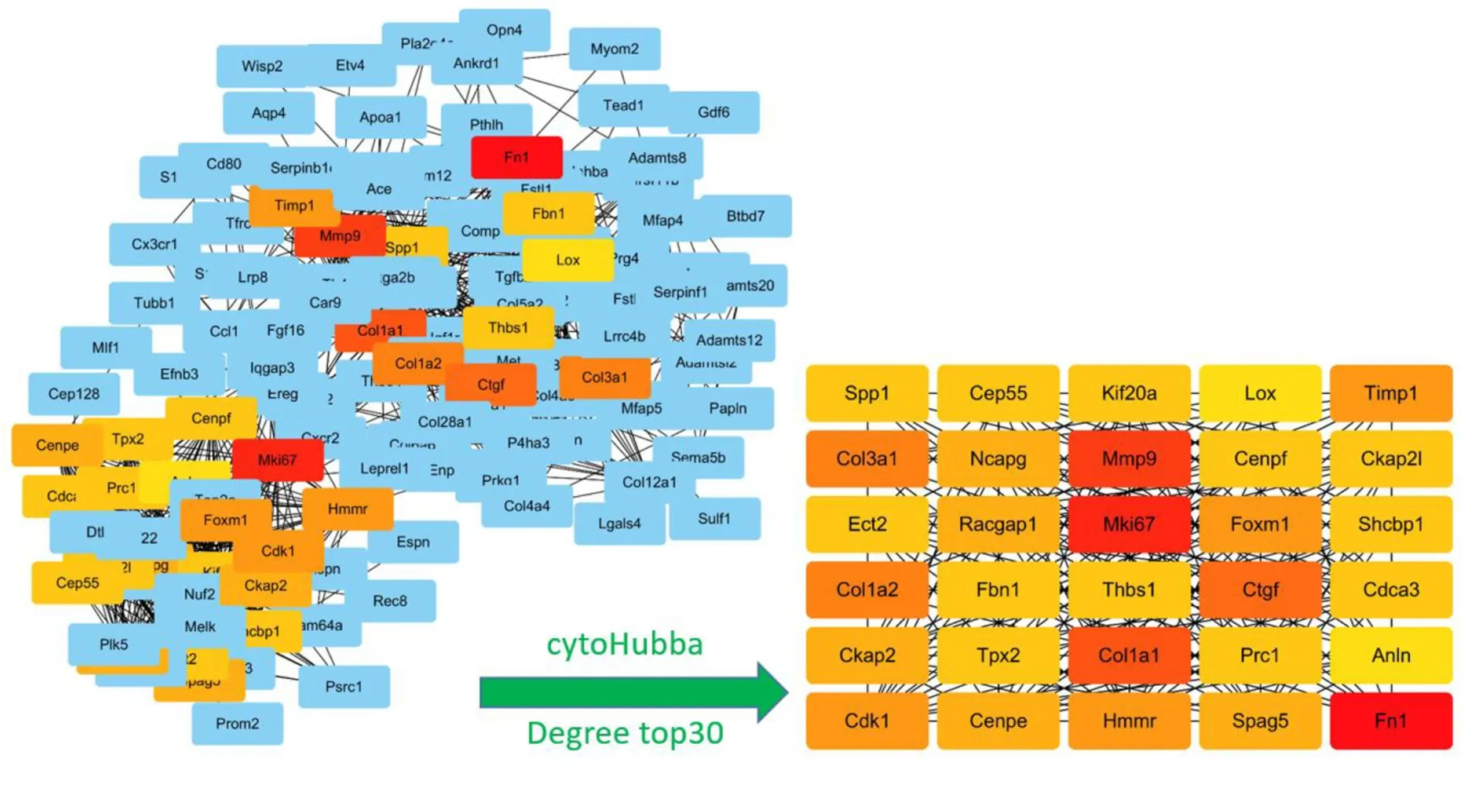
图4 DEGs 构成的PPI 网络中鉴定核心基因
2.4 核心基因富集分析 核心基因GO 分析提示,BP方面,显著富集的子集包括有丝分裂、胞质分裂和细胞骨架依赖性胞质分裂等;CC 方面,核心基因主要参与构成有丝分裂纺锤体、含胶原的细胞外基质和细胞外基质等;在MF 方面,主要参与微管结合、细胞黏附分子结合和赋予拉伸强度的细胞外基质结构成分等。详见图5。

图5 核心基因GO 富集分析部分可视化(气泡图)
核心基因KEGG 富集分析提示,核心基因参与细胞外基质-受体相互作用、黏着斑、糖尿病并发症中的AGE-RAGE、PI3K-Akt、血 小 板 活化、转化生 长 因 子(TGF)-β和p53 等信号通路中。详见图6。
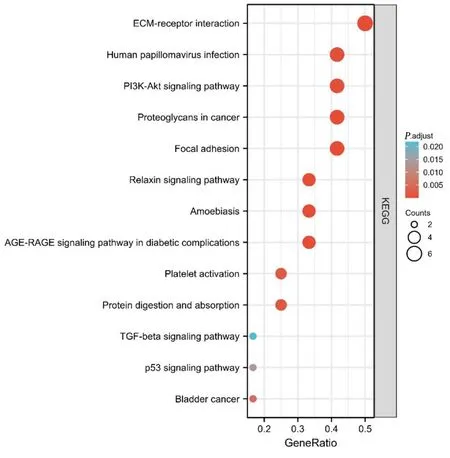
图6 核心基因KEGG 富集分析部分可视化(气泡图)
2.5 核心基因与心力衰竭(舒张性)的相互作用关联性分析 本研究利用CTD 数据库分析核心基因与心力衰竭(舒张性)风险之间的关系。核心基因与心力衰竭(舒张性)的相互作用关联性分析所示,Col1a1、Fn1、Mmp9、Timp1、Spp1、Col3a1、Cdk1 和Thbs1 等与心力衰竭(舒张性)关联性评分较高。详见图7。
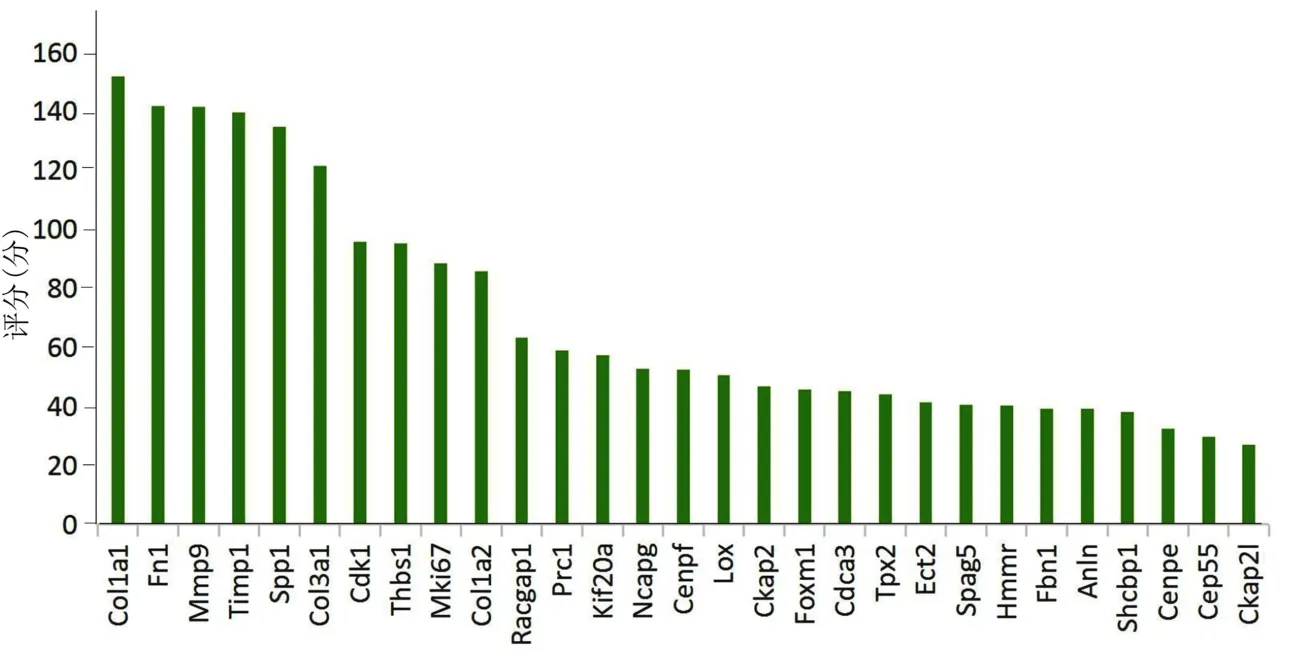
图7 CTD 数据库中核心基因与心力衰竭(舒张性)关联性评分
3 讨 论
HFpEF 是一种表型极其复杂的疾病综合征,涉及炎症、氧化应激、心肌肥厚/纤维化、代谢紊乱等,病理机制研究仍不甚透彻,且缺乏特效治疗。故进一步探索其致病机制有着深远的意义。本研究基于GEO 数据进行生物信息学分析,有助于为疾病的防治提供理论支持。
HFpEF 的发生发展涉及多种基因表达调控失衡。本研究发现HFpEF 小鼠心肌组织中337 个基因表达存在显著性差异。富集分析提示DEGs 在细胞外结构和基质的构成、损伤应答和愈合、胶原纤维构成、血液凝固、细胞趋化性、胶原纤维、糖胺聚糖结合、受体配体活性、纤连蛋白结合和肝素结合等不同GO 子集方面发挥重要作用。KEGG 提示DEGs 参与细胞外基质-受体相互作用、黏着斑、PI3K-Akt、血小板活化、AGERAGE、HIF-1 等。在HFpEF 中,内皮细胞在炎症、应激或损伤刺激下可分泌促炎因子,促进单核细胞黏附、迁移及分化为巨噬细胞。而巨噬细胞可分泌多种介质和促炎细胞因子驱动静止的成纤维细胞转化为增殖和基质合成活跃的肌成纤维细胞,后者持续激活产生细胞外基质蛋白如胶原蛋白等,促使心肌肥大僵硬而出现舒张性功能障碍。本研究分析发现,DEGs 参与这些生物学过程及细胞外基质-受体相互作用、黏着斑信号通路,与HFpEF 病理生理学过程相符合。同时,PI3K-Akt 通路在HFpEF 中激活,PI3K-Akt 在调节心肌细胞生长、心肌血管生成和细胞死亡中起重要作用。胰岛素样生长因子1(IGF1)-PI3K-Akt 信号通路在调节运动诱导的生理性心脏肥大和心脏保护中发挥关键作用[15];增加的PI3K 活性对心脏功能和纤维化有利并削弱病理性生长[16]。也有研究发现,通过主动脉横向缩窄建立的兔舒张性心力衰竭模型中,黏着斑和PI3K-Akt 通路被激活[17];在高脂联合N-ω-硝基-l-精氨酸甲酯(L-NAME)饲养诱导的HFpEF 小鼠模型甲基化研究中,PI3K-Akt 信号通路同样被激活[18],这些进一步证实了本研究分析结果的可靠性及PI3K-Akt 信号通路在HFpEF 发生发展中的重要作用。心力衰竭是一种促血栓状态,心力衰竭病人血小板及血管对一氧化氮(NO)的应答被削弱;与健康个体相比,NO 供体硝普钠对血小板聚集的抑制在HFpEF-心房颤动病人中受损[19]。HFpEF 多伴有肥胖、糖尿病等表型,两者都可能导致炎症和纤维化心房和心室肌病的发展,是全身性血栓栓塞的危险因素[20]。经分析提示HFpEF 小鼠DEGs 参与血液凝固、血小板活化通路,该通路中富集的 DEGs 可能在 HFpEF 血栓事件中起作用。HFpEF 血糖代谢异常,晚期糖化终产物在体内积聚并作用于受体,促进成纤维细胞向肌成纤维细胞的分化,进而触发下游细胞外基质的重塑、炎症和氧化应激等一系列病理过程[21]。富集于AGE-RAGE 信号通路中的DEGs 可能影响HFpEF 的进程。HIF-1α是一种高度保守的转录因子,在组织对缺氧的反应中起关键作用,在肥胖病人的脂肪组织中表达增加,导致胶原蛋白Ⅰ、Ⅲ、Ⅳ和赖氨酰氧化酶等促纤维化转录程序上调;HIF-1α也可介导肥胖相关炎症的M1 巨噬细胞募集,释放炎性细胞因子,并增加胶原蛋白、TGF-β和结缔组织生长因子等的表达。这些综合因素加速心脏纤维化并损害心脏舒张功能;而抑制高脂饮食小鼠脂肪组织中HIF-1α的表达可抑制纤维化并减少脂肪组织中的炎症[22]。本研究发现DEGs 参与HIF-1 通路,提示HIF-1α通路可能是促成HFpEF 的关键环节。
为进一步剖析DEGs 在HFpEF 发病机制中的作用,本研究构建PPI 网络,筛选出核心基因,即Fn1、Mki67、Mmp9、Col1a1、Ctgf、Col1a2、Col3a1、Timp1、Cdk1、Hmmr、Foxm1、Racgap1、Ckap2、Spag5、Ncapg、Cenpe、Fbn1、Spp1、Thbs1、Shcbp1、Cdca3、Ckap2l、Tpx2、Cenpf、Kif20a、Cep55、Prc1、Ect2、Lox 和Anln。并对核心基因进行富集分析。研究表明,持续的压力导致心脏重塑,其中,心肌细胞死亡超过更新,导致进行性心力衰竭;在多数情况下,心肌细胞中的有丝分裂可能代表干细胞的成熟后代;心肌细胞更新可能主要由内源性心脏干细胞和血源性干细胞介导,但这种生物学现象的能力有限[23]。核心基因参与有丝分裂胞质分裂,提示可能为心肌损伤后代偿。核心基因参与细胞外基质、微管结合、细胞黏附分子结合和赋予拉伸强度的细胞外基质结构成分等,与之前的分析符合,提示HFpEF 中存在活跃的结构重塑。与DEGs 通路富集相比,核心基因还参与TGF-β等信号通路中。TGF-β信号通路在心力衰竭发生发展中有着极其重要的作用。在HFpEF 中,TGF-β信号传导参与病理性肥大和纤维化的发展。核心基因Thbs1 和Fbn1 参与该通路。Thbs1 是TGF-β通路激活的关键调节因子,体外实验已证明,miR-221 可靶向抑制Thbs1 蛋白表达,进而抑制TGF-β信号通路的激活,削弱肌成纤维细胞活化、胶原蛋白分泌和心肌纤维化[24],提示该基因可能作为HFpEF 的治疗靶点。Fbn1 家族包括潜在的TGF-β结合蛋白等成员,在体外控制TGF-β1的释放、靶向和激活,并且还作为富含原纤维蛋白的微纤维的结构成分发挥作用[25],在动脉损伤应答中起关键作用,但其在HFpEF 中的作用尚未见报道。
本研究还利用CTD 数据库将核心基因与心力衰竭(舒张性)的相互作用进行关联性分析,关联性评分较高的为Col1a1、Fn1、Mmp9、Timp1、Spp1、Col3a1、Cdk1 和Thbs1 等。这些基因单一或多个同时参与细胞外基质-受体相互作用、黏着斑、PI3K-Akt、血小板活化、AGE-RAGE、血小板活化、HIF-1 等信号通路中,与心力衰竭发生相关。Hua 等[26]发现Col1a1 可能是心力衰竭的血浆生物标志物,与心力衰竭进展相关,且可预测从心力衰竭发病到心脏移植后1 年生存率。通过腹主动脉缩窄构建的小鼠压力过载性舒张性心力衰竭中,氧化还原失衡,基质金属蛋白酶(MMP)-2/9 的表达活性增加,胶原蛋白Col1a1 降解增加,从而诱发心力衰竭;而MMP 对Col1a1 基因的切割位点发生突变后可改善上述病理状态[27]。纤连蛋白(Fn1 编码)和胶原蛋白alpha-1(Ⅲ)链(Col3a1 编码)均属于细胞外基质成分;在压力过载性小鼠模型中使用骨桥蛋白(Spp1 编码)治疗可预防心肌细胞肥大和心脏纤维化,阻断PI3K-Akt 通路磷酸化,降低纤连蛋白和胶原蛋白alpha-1(Ⅲ)链等的表达,预防心力衰竭[28]。金属蛋白酶组织抑制剂1(Timp1 编码)是MMPs 的特异性抑制剂,与MMPs 共同作为细胞外基质组成的关键调节剂,由于其动态活性,可作为HFpEF 心肌纤维化及心脏功能的预后指标[29]。重塑特异性标志物骨桥蛋白(Spp1 编码)可预测HFpEF 病人18 个月时的全因死亡率和/或心力衰竭相关再住院率[30]。氧化应激是HFpEF 的表型之一,其诱导心肌细胞周期停滞,导致心肌细胞丢失;而补充硒剂可通过激活PI3K-Akt 通路使细胞周期蛋白依赖性激酶1(Cdk1 编码)表达增加,削弱氧化应激对心力衰竭的影响。其余核心基因也可能涉及HFpEF 发病机制,值得进一步探讨。
综上所述,本研究基于转录谱数据挖掘进行研究,有助于进一步了解HFpEF 发病机制,为疾病防治及后续科研提供一定的理论支持。但本研究也存在一定局限性,比如未能针对某一功能集或通路内的DEGs 进一步深度探讨,这也是后续研究的方向。另外,HFpEF差异表达基因丰富,DEGs 参与众多生物学功能和/或信号通路,本研究未能针对所有DEGs 及可能涉及的机制展开讨论。
