McCune-Albright综合征2例报道并文献复习*
2022-12-20王静娜王杰英李荣敏雷淑琴桑艳梅
王静娜,王杰英,李荣敏,常 洁,雷淑琴,桑艳梅
[1.保定市儿童医院/保定市儿童呼吸消化疾病临床研究重点实验室,河北保定 071000;2.国家儿童医学中心(北京)首都医科大学附属北京儿童医院/儿科学重点学科内分泌遗传代谢中心]
McCune-Albright综合征(McCune-Albright syndrome,MAS)是一种罕见的体细胞基因突变病,主要由编码鸟苷酸结合蛋白α亚基(alpha-subunit of the Gs stimulatory protein, Gsa)的鸟苷酸结合蛋白a活性刺激肽(guanine nucleoide binding protein alpha stimulating activity polypeptide,GNAS)基因发生突变所致。临床可表现为外周性性早熟、皮肤牛奶咖啡斑及多发性或单发性骨纤维发育不良(Fibrous dysplasia of bone,FD)等。其发病率较低,约为十万至百万分之一[1]。部分病例临床表现不典型,临床极易漏诊。本文对我院收治的2名MAS患儿的临床资料进行了总结分析,并对相关文献进行了复习,以期提高临床医生对该病的认识。
1 临床资料
病例1。一般资料:患儿女性,9个月,因“生长迟缓8个月,阴道出血3个月”于2018年7月25日就诊。患儿出生后家长即发现其左侧上肢、左侧前胸、左侧后背及左侧面颊部存在片状棕色色素沉着,边缘不规则。余无异常,未予重视,后一直身长、体重增长缓慢。6个月时开始出现阴道出血,出血周期家长叙述不清,每次量少,每次持续3~5 d。曾多次到当地医院就诊,未能确诊。入院前4 d,患儿因再次出现阴道出血到我院就诊。个人史:患儿系第1胎第1产,足月剖宫产,无难产、窒息史,出生体重2.7 kg。新生儿期生长1.0 kg,抬头、翻身、爬行、站立、认人与同龄儿无显著差异。母孕期体健,父母非近亲婚配,家族中无类似疾病。
体格检查:体重7.0 kg,身长63.8 cm;面容无特殊;左侧上肢、左侧前胸、左侧后背及左侧面颊部可见不规则牛奶咖啡斑;背部无痤疮、无脂肪垫。甲状腺不大,双侧乳房Tanner B2期,双侧乳核约2.0 cm×2.0 cm,双侧乳晕均无着色;外阴部汗毛较多,大阴唇肥厚,覆盖小阴唇;无腋毛;脊柱四肢无畸形。
实验室检查:血尿常规、肝肾功能、血电解质均正常,甲状腺功能、胰岛素样生长因子-1正常。内分泌相关检测项目及戈那瑞林激发试验结果见表1、2。
影像学检查:(1)乳腺彩超:双侧乳腺较同龄儿增大,左1.6 cm×0.6 cm×1.4 cm,右1.5 cm×0.7 cm×1.4 cm,腺体形态、结构未见异常,未见确切囊、实性占位。(2)肾上腺彩超:双侧肾上腺区未见异常占位性回声及增大的肾上腺。(3)子宫附件彩超:子宫内可见宫内膜,子宫彩色血流丰富(以宫颈显著),阴道内黏稠积液,范围约2.7 cm×1.2 cm×1.7 cm,左侧卵巢1.7 cm×0.6 cm,其内可见卵泡,大者约0.5 cm×0.5 cm;右侧卵巢2.0 cm×0.8 cm,其内可见卵泡,大者约0.7 cm×0.7 cm,右侧卵巢内可见囊性包块,大小约3.8 cm×3.2 cm。印象:卵巢囊肿,子宫形态未见异常。(4)双侧腕关节正位+左膝关节正侧位片:双侧桡骨远端及双胫腓骨远端临时钙化带模糊,建议进一步检查除外代谢性疾病。(5)头颅MRI平扫:双侧基底节区、大脑脚及桥脑背侧对称性异常信号,考虑遗传代谢性疾病。(6)皮肤镜检查:镜下可见色素不规则分布,局部可见皮下血管扩张。
病例2。一般情况:患儿女性,4岁9个月。主因“乳房发育1年4个月,阴道出血2次”于2019年9月8日就诊。患儿3岁6个月时,发现乳房发育,伴触痛,伴阴道出血1次,持续约1 d后出血自行停止,就诊于我院小儿妇科,查子宫彩超示“卵巢囊肿”后就诊于北京儿童医院小儿妇科,予“复幼合剂”口服,乳房停止发育,乳核呈消退趋势,并定期复查随诊。4岁3个月时因转氨酶高于正常,停用复幼合剂。4岁7个月时,再次出现乳房发育,伴触痛,未予治疗。来院前5 d,患儿再次出现阴道出血,量不多,持续4 d停止。近1年身高增长约7 cm。个人史:患儿系第1胎第1产,足月剖宫产,出生史无异常,出生体重2.5 kg。智力及体力发育同正常同龄儿。母孕期体健,父母非近亲婚配,家族中无类似疾病。
体格检查:体重17.5 kg,身高110 cm;面容无特殊,双下肢各见1处浅褐色色素沉着,范围约1.0 cm×0.5 cm,边界不规则;双侧乳房Tanner B2期,双侧乳核约2.0 cm×2.0 cm,轻触痛;双侧乳晕无着色;正常女童外阴,大阴唇覆盖小阴唇,阴毛Tanner PH1期,无腋毛;脊柱四肢无畸形。
实验室检查:血尿常规正常;血电解质、肝肾功能、甲状腺功能五项均正常;肿瘤标志三项正常。内分泌相关检测项目及戈那瑞林激发试验结果见表1、2。

表1 内分泌相关检测项目比较
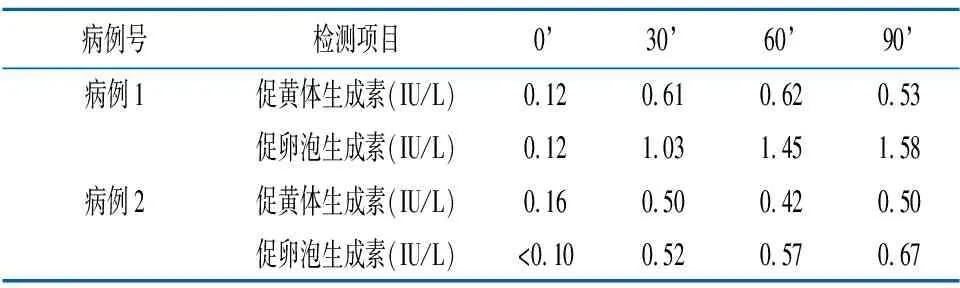
表2 戈那瑞林激发试验
影像学检查:(1)左腕骨正位片:相当于7岁左右(图1)。(2)子宫附件彩超:子宫增大,内膜清晰。子宫长径5.5 cm,横径2.1 cm,宽径1.4 cm。左侧卵巢内探及囊性包块,大小约4.3 cm×2.6 cm,内可见环形膜状回声分层,形成类鸡蛋样结构,透视可。右侧卵巢大小2.1 cm×0.8 cm,卵泡0.7 cm×0.6 cm一枚,0.4 cm×0.4 cm两枚。初步诊断为子宫增大,卵巢囊肿。(3)肾上腺彩超:双侧肾上腺区未见异常占位性回声及增大的肾上腺。(4)双侧尺桡骨正位片:双侧尺桡骨未见明显异常。(5)双侧胫腓骨正位片:右侧胫骨中远段密度欠均匀,远段外侧局部外凸,密度减低,建议CT协诊。(6)双侧胫腓骨CT+三维重建结果显示:右胫骨远端磨玻璃状改变,下部伴环形硬化边——考虑骨纤维发育不良,建议进一步检查除外Albright综合征(图2)。

图1 病例2左腕骨正位片:骨龄相当于7岁左右

图2 病例2双侧胫腓骨CT+三维重建CT片:右胫骨远端磨玻璃状改变,下部伴环形硬化边
2 遗传学检查
测序方法:经监护人知情同意后,对2例患儿进行遗传学检查,分别抽取EDTA抗凝血3 mL。对病例1进行医学全外显子基因捕获panel检测(包含性发育、皮肤、骨骼等相关基因,平均测序深度200×),未发现MAS基因可疑突变位点;又行皮肤活检取患儿牛奶咖啡斑处的皮肤标本,再次进行性腺发育障碍相关基因捕获panel检测(平均测序深度1000×)。对病例2进行性腺发育障碍相关基因捕获panel检测。
基因测序技术路线:提取基因组DNA,将其打断、扩增、建立全基因组文库。应用GenCap液相捕获目标基因技术(北京迈基诺公司),捕获与性腺发育障碍相关的基因的编码外显子区域。利用Illumina Hiseq2000测序仪对捕获到的区域进行双端测序。目标区域测序后,去除测序数据中的接头和低质量数据。运用BWA软件比对到参考基因组上(hg19版本)。用GATK软件对样本的比对数据进行多态性位点的检测,对单核苷酸多态性(single nucleotide polymorphisms,SNPs)和插入缺失突变(InDels)等数据进行统计和分析,查询在千人基因组、ESP6500、ExAC等正常人群数据库中频率,经SIFT、PolyPhen2、MutationTaster、GERP++等数据库进行预测分析。对明确或可能与受检者临床表型相关的基因变异采用Sanger测序进行验证。
基因测序结果: 于病例1患儿皮肤GNAS基因第8外显子区发现了1个c.602G>A杂合错义突变,导致编码产物p.R201H氨基酸变异。见图3、表3。于病例2患儿外周血GNAS基因第1外显子区发现了1个c.46C>T杂合错义突变,导致编码产物p.R16C氨基酸变异。见图4、表3。所有相关检查及治疗患儿监护人均签署知情同意书,并经医院医学伦理委员会批准。

图3 病例1皮肤组织的测序图
携带GNAS c.602G>A杂合错义突变,导致p.R201H氨基酸变异

图4 病例2及父母测序图
病例2携带GNAS基因 c.46C>T杂合错义突变,导致p.R16C氨基酸变异。患儿父亲该位点基因型正常,患儿母亲该位点携带相同的突变。
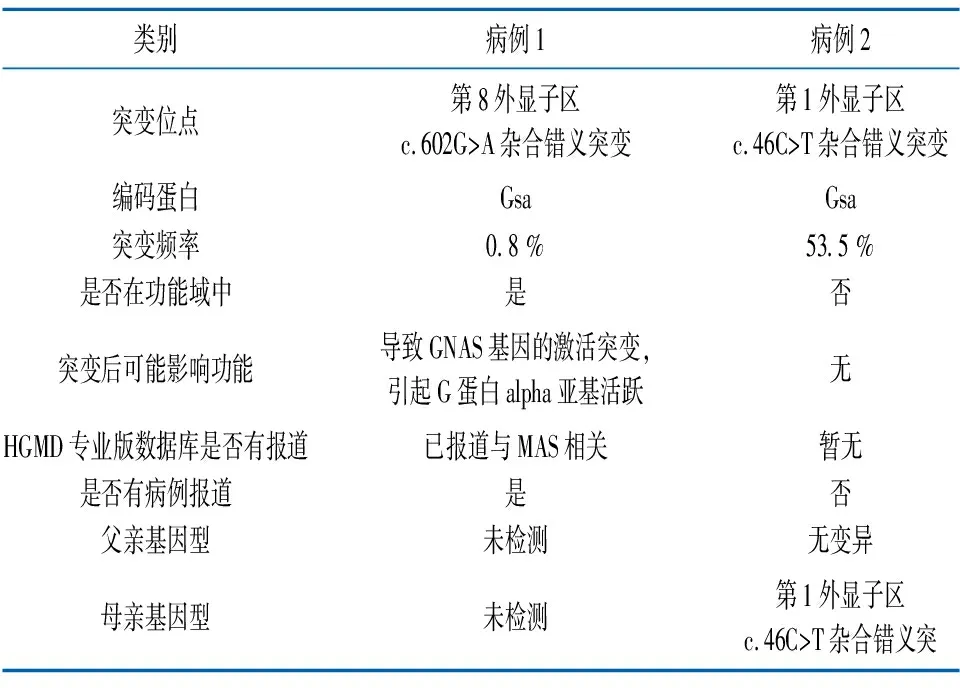
表3 患儿基因测序结果
3 讨论
3.1MAS的发现 MAS是因GNAS基因于胚胎早期发生体细胞突变所致的一种罕见病,由美国医生McCune及Albright分别于1936和1937年首次报道,后被命名为McCune-Albright综合征。男女均可发病,但以女性为多。多于儿童期起病,也有新生儿期起病的报道。
3.2MAS的临床特征 MAS以外周性性早熟、皮肤牛奶咖啡斑及骨纤维发育不良临床三联征为主要表现[2],因受累组织的多样性,临床表现差异较大。性早熟通常是MAS患儿就诊的首要原因,以女孩多见,男女临床表现差异较大。文献报道[3]约85 %的MAS女性患儿有性早熟表现,其中半数以上于4岁前发病。有性早熟表现的患儿中,约半数以上以婴儿期无痛性阴道不规则出血为首要表现,也有初期仅表现为乳房增大(B2-B3期)者。一般情况下,该综合征的乳房增大症状可自行消退。男性患儿性早熟症状相对少见,且比女孩出现时间晚。男孩性早熟主要表现为阴茎增长和继发于睾丸间质细胞增生所致的双侧睾丸增大,也有仅表现单侧睾丸增大者。
在黄珂等[4]的一项研究中,6例MAS患儿均以性早熟为首发表现,起病年龄介于15月龄~8岁8个月,临床主要表现为第二性征提前发育、子宫或卵巢较正常同龄儿大、阴道不规则出血过早出现等。本病出现的性早熟均为外周性性早熟,如不及时诊治,随着病情的进展,可以进一步发展为中枢性性早熟。本研究中,2例患儿分别于6个月和3岁半发病,均表现不规则阴道出血,查体发现均存在第二性征提前发育,盆腔超声均示子宫增大,并可见增大的卵泡、卵巢囊肿,入院后行戈那瑞林激发试验结果显示均为外周性性早熟,符合MAS的临床特征。
FD也是MAS的主要临床表现之一。FD是一种以骨的纤维组织增生、伴骨化不全为特征的良性骨肿瘤。本病骨骼病变出现时间较早,多于儿童期起病。研究资料显示,约90 %MAS患者的FD病灶在3~4岁前即可通过影像学检查被发现,而10岁后则基本不会再出现新的FD病灶[5]。本病患者骨骼X线片上呈典型的“毛玻璃样变”,最常见于颅面骨和股骨近端,也有脊柱、骨盆、长骨骨干、干骺端、甚至骨骺等部位受累者[6]。随着年龄增大及病程的进展,在颅面骨上可表现为病变处骨性隆起,引起面部结构改变而影响美观。而股骨上段可逐渐出现膨胀性弯曲畸形,外观呈典型牧羊人手杖样改变,上述表现被认为是FD最具特色的X线表现。本研究中,病例2确诊时年龄4岁9个月,影像学检查结果显示右胫骨远端磨玻璃状改变,下部伴环形硬化边——考虑骨纤维发育不良,符合MAS骨骼病变的特征。
皮肤牛奶咖啡斑也是MAS三联征表现之一。MAS患儿的牛奶咖啡斑多分布于有骨损害的躯体同侧,一般不穿越身体的中线。患者的皮肤病变可出生时即有,也可于出生后逐渐形成,并可随身体的生长而增长。具体表现为一处或多处边界不规则、点片状大小不等、深黄色或黄棕色改变,多分布于大腿、腰骶部、臀部等处,有时着色较浅,容易被忽略,因其有助于本病的诊断,体检时应仔细寻找。本研究中,病例1左侧上肢、左侧前胸、左侧后背及左侧面颊部均可见不规则牛奶咖啡斑,病例2双下肢各见1处浅褐色色素沉着,范围约1.0 cm×0.5 cm,边界不规则,符合MAS的皮肤病变特征。
除上述典型的三联征表现之外,对于MAS女性患儿,约85 %合并功能性卵巢囊肿,从而导致患儿雌激素水平增高,出现促性腺激素非依赖性性早熟。MAS合并的卵巢囊肿,因常表现为单侧性,易被误诊为恶性肿瘤,致卵巢被误切的可能,临床中须注意鉴别。本研究中,2例患儿初诊时均伴有卵巢囊肿。本综合征伴发的卵巢囊肿大部分具有自限性,不需要手术治疗。只有当卵巢囊肿发生扭转等情况时才考虑手术治疗[7]。
此外,MAS还可表现甲状腺功能亢进症、库欣综合征、甲状旁腺功能亢进症、高泌乳素血症、肢端肥大症、低磷酸盐性佝偻病、以及下丘脑低促性腺激素性性发育不全等内分泌系统的异常[8-10]。部分病例还可有黄疸、肝炎、心律失常、肠息肉等非内分泌系统异常的表现[11-13]。因此,临床中对于确诊为MAS的患者,应该进行全面、细致的检查,以期尽早发现患者伴发的各种异常,及时进行诊治、改善预后。
3.3MAS的致病基因 MAS的致病基因为GNAS,于1991年被Weinstein发现并报道。该基因定位于20q13.3染色体,由13个外显子,12个内含子组成,分子量为20 kb。迄今已发现的导致MAS的GNAS突变均为新生突变,尚未观察到经证实的垂直传播病例。突变类型均为错义突变,尚未发现其他突变类型。已发现的GNAS突变中,最经典的突变是GNAS基因第8外显子区第201号密码子突变,导致第201位的精氨酸被组氨酸或半胱氨酸取代,也有极少数是精氨酸被丝氨酸、甘氨酸或亮氨酸所替代。因GNAS基因的突变为错义点突变,而非生殖细胞突变,故突变的细胞在人体内呈镶嵌式分布,受累严重的组织中含有的突变细胞亦较多,故临床表现呈多样性。有研究资料显示,MAS病人群体中,仅24 %的患儿同时具有三联征的表现,而表现二联征者约占33 %,仅表现一联征者约占40 %[11],因此,仅依据典型临床三联征对本病进行诊断阳性率较低,且易出现漏诊和误诊。临床工作中,对临床表现只有三联征中的一种或者两种的不典型病例,可对GNAS基因进行测序分析以协助诊断[14]。
Lumbroso等[11]对113例MAS患者进行的研究资料显示,MAS三联征患者外周血基因突变检出率为46 %,而仅具有一联征或二联征的患者基因突变检出率则分别降低为8%和21%。该研究对11例患者的骨组织进行基因检测后发现,9例存在GNAS基因突变,检出率高达82 %,提示MAS患者中,组织中也具有较高的基因突变检出率。然而,在这11例患者的皮肤DNA样本中并未检出突变,这再次证实了关于较难在皮肤组织中检出突变的说法[15],可能与在皮肤组织中,作为潜在突变的黑色素细胞所占比例较低有关。
有研究显示,MAS患者牛奶咖啡斑样本中,阳性突变检出率仅27 %。即使有典型三联征的患者,皮肤组织中检出突变者也仅有1/7[11]。有学者曾对患者的皮肤、外周血、胸膜及病变骨组织同时进行基因突变检测,结果仅在患者的骨组织及外周血中检测出了Gsa基因R201C突变[16]。由于MAS的致病基因突变发生在胚胎期,且突变的体细胞克隆随着机体的生长及细胞的分化,散布于不同的组织和器官中,形成突变嵌合体,因此,对MAS病人只进行单一组织的基因检测,容易得出阴性的结果。因此,条件允许的情况下,应从病人的病变部位,如受累的骨骼、卵巢囊肿液细胞或皮肤牛奶咖啡斑处的皮肤等及外周血中同时提取DNA进行突变检测,以期提高Gsa亚基基因突变的阳性检出率。
本研究中,首先对病例1家系的血标本进行了医学全外显子检测(测序深度为200×),结果未能发现与MAS相关的GNAS突变。之后行皮肤活检留取患儿皮肤标本,再次进行MAS相关基因的二代高深度测序(平均测序深度为1000×),结果于GNAS基因第8外显子发现了1个602G>A杂合错义突变,经判定该突变为疑似致病性变异。该突变可导致编码产物第 201 号氨基酸由精氨酸变异为组氨酸,属于经典的MAS致病基因性突变。对于该突变在皮肤组织中突变检出率为0.8 %,属于低频变异,分析原因一是MAS的致病基因突变于皮肤组织中的检出率不高;二是相关检测的深度不够。
本研究于病例2外周血GNAS基因第1外显子区发现了1个c.46C>T杂合错义突变,导致编码产物p.R16C氨基酸变异。该突变在正常人群数据库中的频率为0.00250。经Mutation Taster软件预测结果为有害突变(disease causing)。经GERP++软件预测该突变位点是保守的。该突变此前尚未见文献报道。经家系验证分析,患儿父亲该突变位点基因型正常无变异,患儿母亲该位点携带相同的突变,提示患儿突变来自母亲。因迄今已发现的导致MAS的GNAS突变均为新生突变,父母亲基因型均正常,故目前尚不能判断是否该突变导致患儿发病,有待进一步更深入的研究来阐明。
3.4MAS的治疗策略 MAS迄今尚无有效的根治方法,主要是针对性早熟及骨纤维发育不良进行治疗。对于性早熟,曾采用孕酮、酮康唑等药物治疗,但效果不甚理想。目前抗雌激素治疗药物主要分为两大类:(1)雌激素受体阻断剂:可与雌二醇竞争雌激素受体,进而起到抗雌激素的作用。代表药物为他莫昔芬,一般推荐剂量为20 mg/d,分2次口服。根据不同年龄,剂量可酌减。(2)芳香化酶抑制剂:可抑制雄烯二酮转变为雌酮、睾酮转变为雌二醇,降低体内雌激素的水平。临床常用的药物为来曲唑(第三代芳香化酶抑制剂),推荐剂量为1.5~2.0 mg/m2·d,1次/d口服。对于他莫昔芬治疗效果欠佳者,可考虑改为来曲唑治疗。本研究中,2例患儿性早熟确诊后,均予以他莫昔芬进行了治疗。经随访,阴道出血症状均已消失、乳房发育未再进展。复查盆腔超声,均显示卵巢囊肿较前明显缩小。
多发性骨纤维发育不良的治疗包括手术和非手术治疗。但目前尚缺乏统一的治疗指南,在手术时机及术式的选择上也存在较多争议[17]。大部分患儿的FD病变在跨入青春期及成年后进入相对静止期,只有极少数患儿跨入成年后病变仍持续进展。因此有部分学者提出,对于无症状、进展缓慢的患儿及青少年患者,应严密随访观察,待跨入青春期后或临床症状表现为进行性快速进展时,再依据具体情况采取相应的治疗措施[18]。然而,亦有学者提出应积极切除病变并行预防性视神经管减压,以防止可能发生的神经功能受损和严重的颌面部畸形[19]。非手术治疗中,补充维生素D及钙剂为治疗的基础。对于四肢的FD,特别是下肢骨,加强所在骨肌肉功能训练,尤其是游泳、骑车等不负重的功能训练,可减少骨折发生的风险,也是重要的治疗手段。本研究病例2存在下肢骨骨损害,但由于患儿病变较轻,故暂未予手术治疗,建议其加强功能锻炼,长期随访。
3.5MAS的预后 MAS的预后差异较大,因内分泌异常的程度、范围及病变骨受累的部位、程度而异。外周性性早熟第二性征的变化并非呈进行性加重趋势,其预后比中枢性性早熟要好。而对于骨病损,如发生在上、下颌骨,可导致上呼吸道梗阻,危及生命,如发生在脊柱,可导致神经受压症状。长期随访资料显示,部分患儿成年后可获得生育能力。对于MAS患儿,保持长期监护及恰当处理,大部分预后良好,但也有极少数转化为中枢性性早熟或其他类型的内分泌功能异常,或颅底骨FD增生、硬化致颅神经受压者,预后不良。
MAS为儿童罕见病,发病年龄较早,易造成多系统受损。目前临床三联征表现仍为本病诊断的主要依据,而基因诊断也越来越受到重视。但单一组织基因突变检出率低,如条件允许,应同时进行病变组织多部位基因检测,以协助诊断及早期干预。同时,对于青春期前儿童出现阴道出血情况,应警惕是否存在性发育异常疾病,以避免漏诊和误诊。
