逆流色谱法分离纯化蒲公英中多酚类化合物
2022-12-19高洁宋祥云马天宇刘峰ROMANKachan王晓
高洁,宋祥云,马天宇,刘峰,ROMAN Kachan,王晓*
(1. 齐鲁工业大学(山东省科学院) 山东省分析测试中心,山东 济南 250014;2.齐鲁工业大学(山东科学院) 基辅学院,山东 济南 250353;3.基辅国立工艺设计大学,乌克兰 基辅 01011)
蒲公英为菊科多年生草本植物,具有清热解毒、消痈散结的功效[1]。作为一种药食同源的植物,蒲公英在我国种植广泛,资源丰富。蒲公英的化学成分复杂,主要包括多酚类、萜类、植物甾醇类、倍半萜内酯类和香豆素类等[2]。研究表明,蒲公英多酚类物质具有抗氧化[3]、抗癌[4]、抑菌[5]、抗炎[6]、降血糖[7]等方面的药理活性。
为进一步深入研究蒲公英多酚化合物的药理活性,需要分离制备出更多的单体化合物。蒲公英多酚类物质成分复杂,且分离制备难度大,目前常用的分离制备方法以柱色谱为主[8],其样品制备效率低、耗时长。而逆流色谱具有制备量大、分离效果好、操作方便等特点,其独特的液液分配原理,避免了固体载体对成分的不可逆吸附和降解作用,已被广泛应用于多酚类[9]、生物碱类[10]、三萜类[11]和多肽类[12]等天然产物的制备分离。现阶段采用逆流色谱对蒲公英多酚进行分离的研究报道相对较少[13]。本文以蒲公英药材为原材料,采用pH区带逆流色谱(pH-zone-refining counter-current chromatography,pH-ZRCCC)结合高速逆流色谱(high-speed counter-current chromatography ,HSCCC)对蒲公英多酚进行分离纯化,并对其化学结构进行鉴定,为蒲公英药理活性和质量标准的研究提供技术支持。
1 材料与方法
1.1 主要原料
蒲公英购于山东建联盛嘉中药有限公司中药饮片厂,经齐鲁工业大学药学院刘伟教授鉴定为菊科植物蒲公英的干燥全草;实验用正丁醇、正己烷、甲醇、无水乙醇、乙酸乙酯、石油醚、氨水、三氟乙酸等试剂均为分析纯,购于天津市富宇精细化工有限公司;用于HPLC分析的甲醇为色谱纯,购于美国天地公司。
1.2 仪器设备
TBE-300C高速逆流色谱仪、TBP-5002输液泵、DC-0506低温恒温槽(上海同田生物技术股份有限公司),8823B紫外检测器(北京宾达英创科技有限公司),3057-11便携式记录仪(重庆川仪自动化股份有限公司),UB-7 pH计(丹佛仪器有限公司),BUCHI旋转蒸发仪(瑞士BUCHI公司),Agilent 1120型高效液相色谱仪(美国安捷伦公司),布鲁克ADVANCE DPX 400核磁共振波谱仪、布鲁克AVIII HD 600核磁共振波谱仪(瑞士布鲁克公司)。
2 实验方法
2.1 蒲公英多酚的提取
2.2 目标化合物在两相溶剂系统中分配系数的测定
2.2.1 pH-ZRCCC分配系数的测定
称取约5 mg样品置于10 mL试管中。配置好溶剂系统,取上下相各5 mL置于装有样品的试管中,滴加氨水至pH为10左右,震荡试管至样品完全溶解,静置分层。取上下相各10 μL,分别用高效液相色谱(high performance liquid chromatography, HPLC)进行检测。用上相的峰面积(AU)比下相的峰面积(AL)计算碱性条件下样品的分配系数Kbase,计算公式如下:Kbase=AU/AL。再向该试管中滴加三氟乙酸至pH约为2,震荡摇匀,静置分层,按同样的方法各取10 μL上下相,利用HPLC计算分配系数Kacid。
2.2.2 HSCCC分配系数的测定
称取约5 mg样品于10 mL试管中。配置好溶剂系统,取上下相各5 mL于装有样品的试管中,剧烈震荡至样品完全溶解,静置分层,分别取上下相各10 μL利用HPLC进行检测,上相的峰面积(A1)比下相的峰面积(A2)即为分配系数K,计算公式如下:K=A1/A2。
2.3 两相溶剂系统的制备
2.3.1 pH-ZRCCC溶剂体系的配制
称取样品,取等量酸化的上相与不加碱的下相溶解样品,用于后续逆流色谱分离。
2.3.2 HSCCC溶剂体系的配制
称取样品,取等量的上下相将样品溶解备用。
2.4 逆流色谱分离
2.4.1 pH-ZRCCC分离
以30 mL/min将上相泵入高速逆流色谱仪作为固定相,待固定相充满色谱螺旋管后,将样品溶液注入分离柱内。开启速度控制器,使分离柱按顺时针方向以800 r/min的速度旋转,同时以2 mL/min的速度泵入下相溶液。开启紫外检测器,将波长调为280 nm,根据色谱图收集色谱峰组分,并使用pH计检测馏分的酸碱度。分离结束后,使用压力泵将柱内剩余液体吹出,收集于量筒中,用尾吹中固定相的体积比尾吹总体积来计算固定相的保留率。
2.4.2 HSCCC分离
以30 mL/min速度泵入上相作为固定相,待固定相充满色谱螺旋管后,开启速度控制器,使分离柱按顺时针方向以800 r/min的速度旋转,同时以2 mL/min的速度泵入下相。当体系达到流体动力学平衡后,将样品注入分离柱内。开启紫外检测器,在波长280 nm下根据色谱图收集色谱峰组分。分离结束后,使用压力泵将柱内剩余液体吹出,收集于量筒中,用尾吹中固定相的体积比尾吹总体积来计算固定相的保留率。
将分离出的各部分组分减压浓缩至干燥,密封保存于冰箱内。
2.5 HPLC分析和结构鉴定
将蒲公英总样以及分离纯化所得的组分使用HPLC进行分析。HPLC的分析条件按表1进行,色谱柱为Waters C18色谱柱(250 mm×4.6 mm,5 μm),流动相为甲醇(A)-0.1%甲酸水溶液(B),流速1.0 mL/min,检测波长280 nm,进样量10 μL。并采用归一化的方法计算样品纯度。将分离纯化的各组分用二甲基亚砜(dimethyl sulfoxide, DMSO)溶解,经电喷雾质谱(electrospray ionization ion trap mass spectrometry, ESI-MS)和核磁共振波谱法(nuclear magnetic resonance spectroscopy, NMR)分析,鉴定其化学结构。
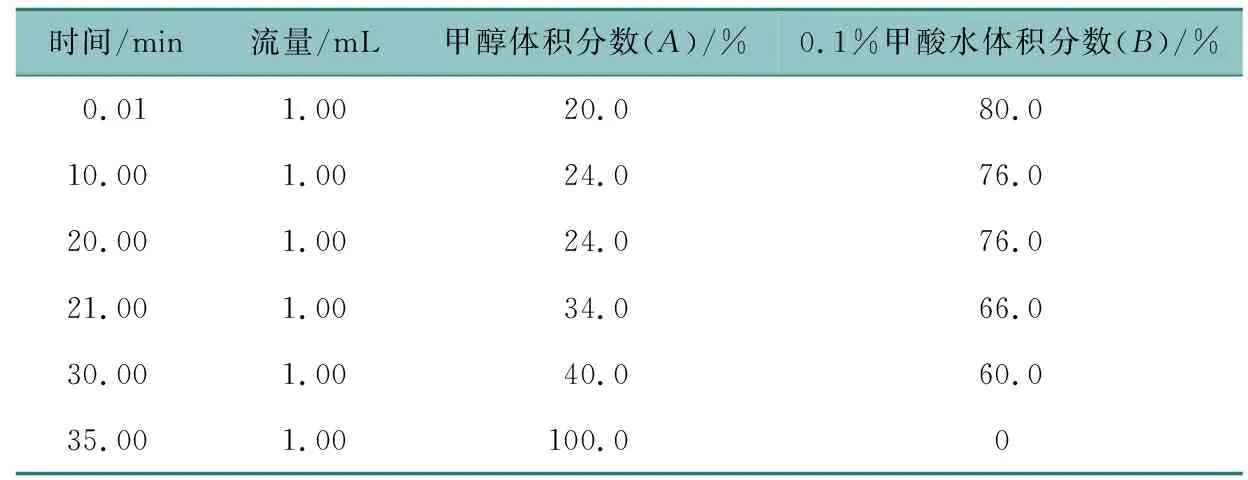
表1 蒲公英HPLC梯度洗脱条件
3 结果与分析
3.1 HPLC分析结果
图1为蒲公英多酚粗提物和制备的纯品的HPLC分析图。在280 nm下使用峰面积归一化法计算,目标化合物I~VI在总样中的峰面积比分别为,40.01%(化合物I),9.07%(化合物II),6.68%(化合物III),1.34%(化合物IV),4.50%(化合物V),8.37%(化合物VI)。
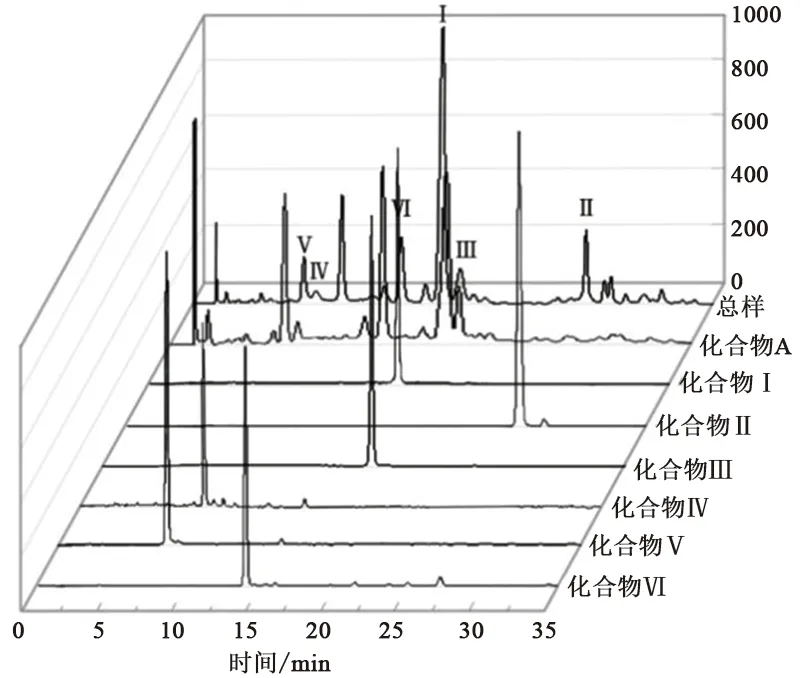
图1 蒲公英多酚粗提物和纯组分的HPLC分析图Fig.1 HPLC analysis of crude extract and pure components of Herba Taraxaci polyphenols
3.2 pH-ZRCCC的分离
pH-ZRCCC制备量大,适用于浓度大于0.1 mmol,最好高于1 mmol的组分[14]。因此首先选用pH-ZRCCC法对蒲公英中含量较高的多酚组分进行分离,同时将微量组分富集。
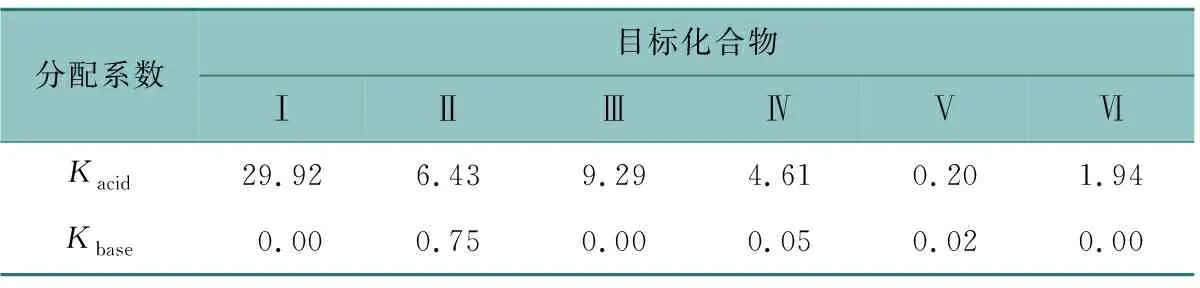
表2 目标化合物I~Ⅵ在溶剂系统中的分配系数Kacid和Kbase

图2 蒲公英粗提物的pH-ZRCCC分离图Fig.2 pH-ZRCCC separation diagram of crude extract of Herba Taraxaci
3.3 HSCCC的分离
混合物A经pH-ZRCCC法分离后未能达到分离的效果,推测其酸碱性可能相近或组分含量较少,适合应用HSCCC法对其再次进行分离。

图3 蒲公英回收组分HSCCC分离图Fig.3 HSCCC separation diagram of recovered components of Herba Taraxaci
3.4 化合物的结构鉴定
化合物I:ESI-MS,m/z:179.0332[M-H]-。1H-NMR(400 MHz,DMSO)δ:8.43(s,1H),7.33(d,J=15.8 Hz,1H), 7.02 (s, 1H), 6.90 (d,J=8.1 Hz, 1H), 6.75 (d,J=8.1 Hz, 1H), 6.18 (d,J=15.8 Hz, 2H)。13C-NMR(100 MHz, DMSO) δ 169.35 (s), 148.64 (s), 146.30 (s), 143.50 (s), 126.40 (s), 121.16 (s), 117.61 (s), 116.38 (s), 115.06 (s)。与文献[17]报道的咖啡酸数据一致,故鉴定化合物Ⅰ为咖啡酸。
化合物II:ESI-MS,m/z:163.0288[M-H]-。1H NMR (400 MHz, DMSO) δ:7.49 (t,J=11.7 Hz, 3H), 6.79 (d,J=8.5 Hz, 2H), 6.29 (d,J=15.9 Hz, 1H), 1.23 (s, 1H)。13C NMR (100 MHz, DMSO) δ 159.88 (s), 144.27 (s), 130.48 (s), 116.20 (s)。与文献[18]报道的对羟基肉桂酸数据一致,故鉴定化合物Ⅱ为对羟基肉桂酸。
化合物III:ESI-MS,m/z:253.0653[M-H]-。1H NMR (400 MHz, DMSO) δ 7.49 (d,J=15.9 Hz, 1H), 7.05 (s, 1H), 7.00 (d,J=8.2 Hz, 1H), 6.77 (d,J=8.1 Hz, 1H), 6.26 (d,J=15.9 Hz, 1H), 4.15 (dd,J=11.2, 4.0 Hz, 1H), 4.00 (dd,J=11.1, 6.5 Hz, 1H), 3.75-3.66 (m, 1H), 1.24 (s, 2H)。13C NMR (100 MHz, DMSO) δ 167.09 (s), 148.92 (s), 146.08 (s), 145.55 (s), 125.96 (s), 121.80 (s), 116.24 (s), 115.23 (s), 114.46 (s), 69.90 (s), 66.08 (s), 63.17 (s)。与文献[19]报道的1-O-咖啡酰基甘油数据一致,故鉴定化合物Ⅲ为1-O-咖啡酰基甘油。
化合物IV:ESI-MS,m/z:167.0277[M-H]-。1H NMR (600 MHz, DMSO) δ 8.84 (s, 1H), 6.64 (d,J=2.6 Hz, 2H), 6.63 (s, 1H), 6.48 (dd,J=8.0, 2.1 Hz, 1H), 3.32 (s, 3H)。13C NMR (150 MHz, DMSO) δ 173.77 (s), 145.43 (s), 144.43 (s), 126.32 (s), 120.46 (s), 117.13 (s), 115.79 (s)。与文献[20]报道的3,4-二羟基苯乙酸数据一致,故鉴定化合物Ⅳ为3,4-二羟基苯乙酸。
化合物V:ESI-MS,m/z:153.0089[M-H]-。1H NMR (400 MHz, DMSO) δ 9.46 (s, 2H), 7.33 (s, 1H), 7.31-7.25 (m, 1H), 6.78 (d,J=8.2 Hz, 1H)。13C NMR (100 MHz, DMSO) δ 167.77 (s), 150.45 (s), 145.34 (s), 122.34 (s), 122.14 (s), 117.01 (s), 115.60 (s)。与文献[21]报道的原儿茶酸数据一致,故鉴定化合物Ⅴ为原儿茶酸。
化合物VI:ESI-MS,m/z:151.0288[M-H]-。1H NMR (400 MHz, DMSO) δ 12.15 (s, 1H), 9.27 (s, 1H), 7.03 (d,J=8.3 Hz, 1H), 6.69 (d,J=8.3 Hz, 1H)。13C NMR (100 MHz, DMSO) δ 173.59 (s), 156.50 (s), 130.71 (s), 125.60 (s), 115.48 (s)。与文献[22]报道的对羟基苯乙酸数据一致,故鉴定化合物Ⅵ为对羟基苯乙酸。
4 结论
本研究利用pH-ZRCCC和HSCCC相结合的方法对蒲公英多酚进行分离,成功地分离出了6个多酚类化合物。在实验过程中首先利用pH-ZRCCC将蒲公英中含量高的多酚类成分分离出来并对低含量成分进行富集,再利用HSCCC实现其他低含量多酚成分的分离,有效地缩短了分离制备的时间。本方法操作简单,快速高效,为分离多酚类化合物提供了新的高效制备方法,同时也为蒲公英的开发利用提供了技术支撑,具有较好的应用价值。
