基于线粒体和初级共生菌基因序列的荻草谷网蚜6个地理种群的遗传多样性研究
2022-12-19孙靖轩谭晓玲FRANCISFrric陈巨莲
孙靖轩,李 迁,谭晓玲,范 佳,张 勇,FRANCIS Frédéric,陈巨莲*
(1.中国农业科学院植物保护研究所,植物病虫害生物学国家重点实验室,北京 100193;2.比利时列日大学让布鲁农业生物技术学院功能与进化昆虫学系,让布鲁 5030;3.黑龙江省农业科学院牡丹江分院,牡丹江 157000)
荻草谷网蚜Sitobionmiscanthi(Takahashi)是我国一种严重为害小麦的害虫,在我国一直被广泛误用为麦长管蚜Sitobionavenae(Fabricius),1999年由我国知名蚜虫分类专家张广学主编的《西北农林蚜虫志》在中国环境科学出版社正式发表,对该蚜虫学名进行勘误校正。麦长管蚜在我国境内仅存在于新疆伊犁等少数地区,其腹管长度和触角节次生感觉圈分布位置与荻草谷网蚜存在一定差异[1]。本课题组通过对基因组研究也验证了我国麦蚜优势种为荻草谷网蚜[2]。荻草谷网蚜原产于亚洲,主要分布于中国、印度以及太平洋地区[3],每年暴发,对小麦的安全生产造成严重威胁。目前,我国研究报道中,包括本文引用的参考文献中“麦长管蚜”即为“荻草谷网蚜”。该蚜虫在南方和北方均具有一定迁飞性[4]。了解其种群遗传多样性和相关因素有助于推测其迁飞路径并为害虫防治提供理论基础[5]。早期的迁飞研究以传统的越冬调查、灯光诱集、雷达监测等测报方法来预报虫情。张向才等依据越冬调查的结果推测麦长管蚜在5月份可以随西南气流由豫西、晋南、关中、陇南、陇东和延安等北方冬麦区迁往银川、内蒙古、张家口、承德等春麦区[6]。国伟等推测荻草谷网蚜种群南北向的交流要强于东西向[7]。董庆周等认为宁夏银川存在外来蚜源远距离迁入的现象,且外来蚜源可以成为春季田间荻草谷网蚜种群的主体[8]。了解麦蚜虫源及迁飞路径是准确预测预报和有效防治的基础。然而,由于蚜虫个体小,缺少合适研究技术方法,因此荻草谷网蚜的迁飞路径及迁飞规律的研究一直进展缓慢。
近年来分子生物学技术高速发展为蚜虫迁飞研究提供了新的技术手段。研究蚜虫群体遗传变异的地理分布可以推断不同地区蚜虫的遗传多样性和可能的迁飞路线,为研究蚜虫的迁飞扩散等提供分子遗传学证据[9]。其中线粒体DNA以进化速率快,遗传多样性丰富等特点,在昆虫近缘种和地理种群进化研究中得到广泛应用[10]。而蚜虫初级共生菌作为近期研究热点,其对于蚜虫营养代谢和正常发育至关重要,并且与蚜虫具有专性共生关系,利用共生菌相关基因作为昆虫分子标记的补充可以解决分子系统进化及种下不同地理种群分化等问题[11-13]。因此,本研究选用线粒体细胞色素C氧化酶亚基Ⅰ(COⅠ)和初级共生菌Buchneraaphidicola的两个基因(gnd和trpA)对我国6个荻草谷网蚜地理种群的遗传多样性、种群结构和种群历史动态进行探究,分析其系统发育关系。旨在揭示在地理隔离和相关的生态因素影响下的荻草谷网蚜不同种群的遗传结构特征,以期为该蚜虫的迁飞扩散路径提供分子生态学依据。
1 材料与方法
1.1 样本采集
从6个地点(表1)共收集了316头荻草谷网蚜成虫个体,涵盖了中国大部分主要麦区。所有样本均储存在-20℃的无水乙醇中,保存后提取DNA。
1.2 DNA提取和测序
使用TIANamp基因组DNA试剂盒(天根生物科技有限公司,北京)提取单头成虫的基因组DNA。以线粒体蛋白编码基因(COⅠ的部分序列)[14]和2个共生菌基因(gnd和trpA)[15-16]作为分子标记(表2)。PCR扩增体系为25 μL,包括DNA模板1 μL (45 ng/μL),10×PCR buffer 2.5 μL,上下游引物各0.5 μL,TaqDNA聚合酶1 μL,ddH2O 17.5 μL。反应条件为:94℃预变性0.5~1 min;94℃变性30 s,退火30 s(退火温度见表2),72℃延伸1 min,35~40个循环;72℃延伸10 min。PCR产物用1.0%琼脂糖凝胶检测,在紫外光下对凝胶进行照相并记录结果。纯化的PCR产物进行双向测序(三博生物技术有限公司,北京)。
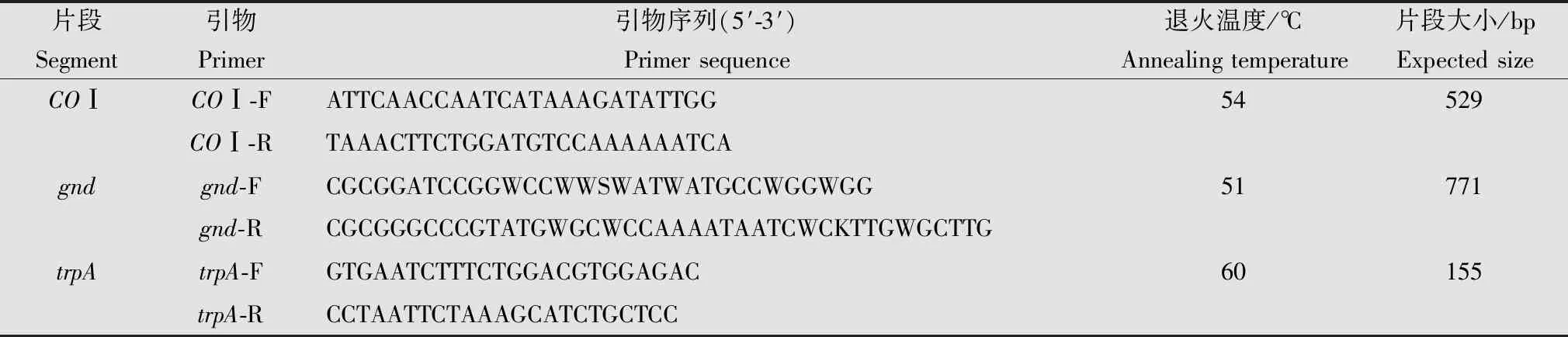
表2 试验所用4对标记序列引物1)Table 2 Four pairs of primers for marker sequences
1.3 种群结构和遗传多样性
拼接校对后的序列使用MEGA 7.0中的Clustal W[17]进行比对,比对时采用默认参数。使用DnaSP 5.0计算多态位点数(S)、单倍型数(Ht)、单倍型多样性(Hd)、核苷酸多样性(π)和平均核苷酸差异数(K),进而推算各地种群遗传多样性高低。使用Arlequin计算两两地理种群之间的成对分化指数FST,来估计种群间的遗传差异,计算所有种群的成对FST值[18-19]。成对FST通常用于衡量种群间的遗传分化程度,不同基因的成对FST值差异较大,其取值范围在0~1之间。FST≤0.05表示两组间几乎没有遗传分化,0.05
1.4 种群系统发育分析
通过MEGA 7.0软件,利用邻接法(neighbor joining,NJ)构建基于Kimura双参数模型(Kimura-2-parameter,K2P)[20]的系统发育树,重复1 000次bootstrap检验。然后,使用PopART 1.7[21]构建单倍型网络中介图(median-joining networks),网络图可以用来揭示单倍型之间的关系。单倍型网络图呈放射状分布,祖先单倍型一般应位于网络图的中间,并在地理区域内广泛分布,最新出现的单倍型位于网络图的边缘,其地理分布范围非常有限。
1.5 种群遗传结构和种群历史动态分析
使用SAMOVA 1.051[22]进行分子空间变异(spatial analysis of molecular variance,SAMOVA)分析,检测种群间最大的遗传障碍。设置组数K为2~5,并进行100次置换检验。当种群内的遗传分化最小(FSC最小),组间遗传分化最大(FCT最大)或者达到平台时的分组结果(即给出的K值)为最优的分组结果。利用Arlequin计算Fu’sFs和Tajima’sD,检验种群是否偏离了中性进化模型,以了解各种群的虫口动态历史。当Fu’sFs和Tajima’sD值为显著的负值时表明种群经历了种群扩张;当Fu’sFs接近于0时表明指定种群为稳定的种群;当Fu’sFs为显著的正值时表明种群可能经历了瓶颈效应或者出现了种群分化。
2 结果与分析
2.1 种群遗传多样性分析
荻草谷网蚜6个种群中的3个不同基因COⅠ、gnd和trpA遗传变异分析结果表明,不同种群遗传结构存在明显差异,在316头个体中存在33个COⅠ单倍型、6个gnd单倍型和19个trpA单倍型(表3)。不管是COⅠ、gnd,还是trpA标记,银川和泰安种群中都含有特有单倍型个体。COⅠ的核苷酸多样性最高。发现了34个多态位点,其中包括20个简约信息位点和14个单变量位点,分别占总数的3.78%(20/529)和2.65%(14/529)。

表3 不同地理种群荻草谷网蚜的序列变异Table 3 Sequences variation in different Sitobion miscanthi geographical populations
进一步分析表明,在所有种群中,COⅠ序列多态位点数(S)为每种群6~24个(表4),单倍型数(Ht)为5~14个,单倍型多样性(Hd)为0.449~0.983,平均核苷酸差异数(K)为1.865~6.625,核苷酸多样性(π)为0.003 53~0.012 52。武汉种群的多态位点数量最多(24个),单倍型多样性(Hd)最高(0.983),单倍型数量(Ht)也最多(14个)。同时,武汉和银川种群表现出较高的单倍型多样性(Hd)分别为0.983、0.894、核苷酸多样性(π)分别为0.012 52、0.008 05和平均核苷酸差异数(K)分别为6.625、4.258。通过分析COⅠ标记的遗传多样性参数,可以看出蚜虫两两种群之间存在着明显的多态性差异,武汉和银川种群的遗传多样性较高。
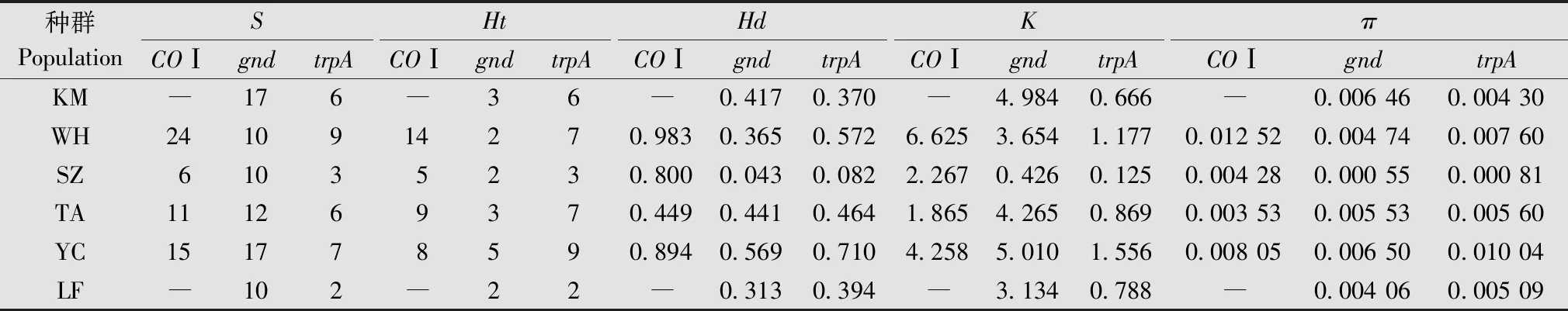
表4 荻草谷网蚜的COⅠ、gnd和trpA遗传多样性指数1)Table 4 Genetic diversity indices of Sitobion miscanthi based on COⅠ,gnd and trpA
gnd序列的遗传多样性分析表明,6个种群的序列多态位点数(S)为10~17个,单倍型数(Ht)为2~5个(表4)。银川显示出最高的单倍型多样性(Hd)为0.569、核苷酸多样性(π)为0.006 5和平均核苷酸差异数(K)为5.01。昆明和银川显示出大量的突变位点(S)共计17个。该分子标记含有大量可变位点,但单倍型的数量很少。
trpA序列的遗传多样性分析表明,6个种群的序列多态位点数(S)为2~9个(表4),单倍型数(Ht)为2~9个。单倍型多样性(Hd)为0.082~0.71,核苷酸多样性(π)为0.000 8~0.01,平均核苷酸差异数(K)为0.125~1.177。武汉种群的多态位点数量最多,有9个,银川种群的单倍型最多。银川和武汉种群遗传多样性较高,苏州种群遗传多样性较低。
2.2 种群间遗传结构
基于COⅠ的成对分化指数FST值在0.115 79~0.903 54的范围内(表5)。大多数成对分化指数FST大于0.25,表明种群高度分化。然而,武汉种群与廊坊种群表现出中等程度的遗传分化(FST=0.115 79)。武汉与苏州种群和泰安种群分别表现出较高的遗传分化,其FST值分别为0.240 64、0.152 21。

表5 基于COⅠ线粒体基因的荻草谷网蚜种群的成对分化指数FSTTable 5 Pairwise FST values of Sitobion miscanthi populations based on the mitochondrial gene COⅠ
基于gnd基因的成对FST在0.000 75~0.701 48的范围内(表6)。苏州与廊坊种群间也表现出中等程度的遗传分化(FST=0.139 61)。苏州种群与武汉(FST=0.169 93)、昆明(FST=0.178 49)和泰安(FST=0.206 63)3个种群间都表现出较高的遗传分化。银川种群与武汉、昆明、泰安、廊坊等5个种群分别表现出高度的遗传分化(FST值分别为0.701 48、0.436 82、0.381 06、0.376 63和0.475 21)。廊坊种群分别与武汉、昆明和泰安种群几乎没有遗传分化(FST分别为0.024 22、0.010 05和0.000 75)。泰安种群与武汉、昆明种群几乎没有遗传分化(FST分别为0.012 09、0.003 40)。武汉和昆明种群之间也几乎没有表现出遗传分化(FST=0.010 76)。

表6 基于gnd基因的荻草谷网蚜种群的成对分化指数FSTTable 6 Pairwise FST values of Sitobion miscanthi populations based on the mitochondrial gene gnd
基于trpA基因的成对FST值范围为0.013 25~0.464 79(表7)。部分成对FST小于0.05,表明种群之间几乎没有遗传分化。银川种群与苏州和昆明种群的遗传分化程度很高(FST分别为0.464 79、0.304 75),与武汉、泰安和廊坊种群的遗传分化指数FST分别为0.159 70、0.210 46、0.180 28。苏州与廊坊种群之间也表现出较高的遗传分化(FST=0.190 97)。苏州种群与武汉、泰安两种群的成对FST值表现出中等程度的遗传分化(FST分别为0.130 79、0.120 19)。

表7 基于trpA基因的荻草谷网蚜种群的成对分化指数FSTTable 7 Pairwise FST values of Sitobion miscanthi populations based on the mitochondrial gene trpA
SAMOVA对线粒体基因(COⅠ)和初级共生菌gnd和trpA分子标记的分析结果显示:COⅠ基因的分析结果如(图1a)所示,即当K=3时,FCT显著升高且达到最大值(FCT=0.364,P<0.001)。K=3时,银川和廊坊为第一组;苏州为第二组;剩余的3个种群为第三组。
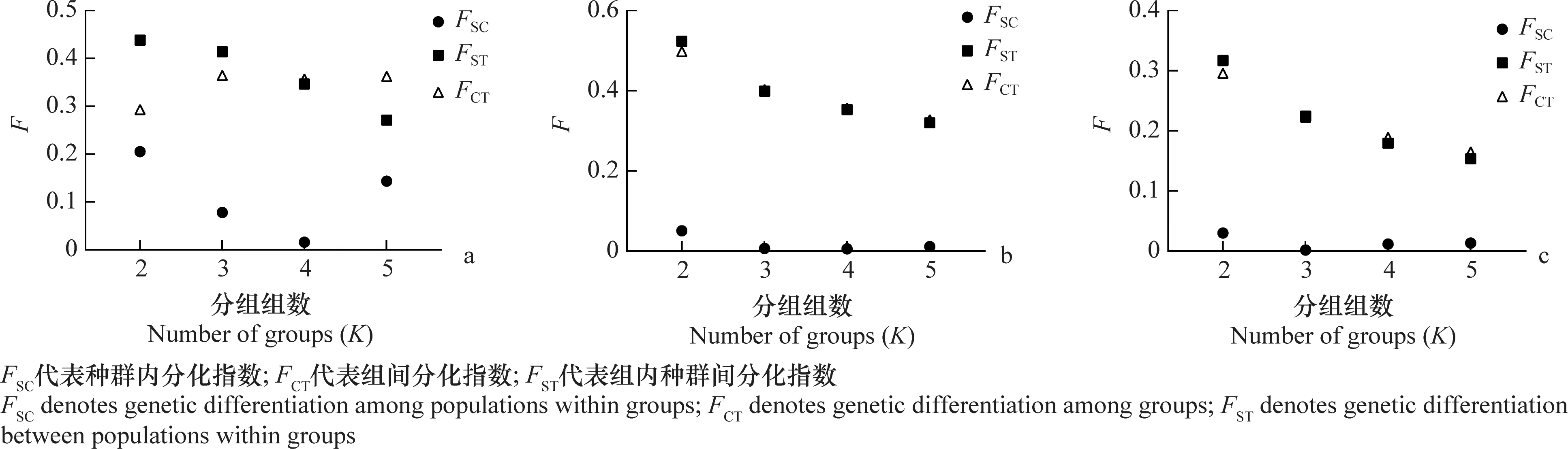
图1 基于COⅠ (a)、gnd (b)和trpA (c)分子标记的遗传分化指数(F)随着分组组数(K)的变化Fig.1 Changes of genetic differentiation indices with K values by SAMOVA analysis based on COⅠ (a),gnd (b),and trpA (c) markers
gnd基因的分析如图1b所示,FCT值从K=2到5呈现下降趋势。K=3时,银川为第一组;苏州为第二组;剩余的4个种群为第三组。
trpA基因的分析如图1c所示,FCT值从K=2到5呈现缓慢下降趋势。K=3时,银川为第一组;苏州为第二组;剩余的4个种群为第三组。该结果与gnd基因的划分结果一致。
结合以上3个基因单独划分的SAMOVA结果,我们采用K=3作为划分种群最佳分组数量。尽管这些群体并没有产生完全相同的群组划分,但苏州和银川种群与其他4个种群不同。
总之,通过对3个基因片段在两两种群间成对分化指数的分析显示结果和SAMOVA的分析结果基本一致,即荻草谷网蚜在苏州、银川以及其他种群3个不同区域间存在着明显的遗传分化,同时各个区域内部存在着较为频繁的基因交流。
2.3 单倍型系统发生
使用邻接法构建了荻草谷网蚜的3个分子标记单倍型的系统发育树。每个进化树选择同属蚜虫苦苣无网蚜Acyrthosiphonlactucae、蓝苜蓿蚜Acyrthosiphonkondoi等相关序列作为外群。
线粒体单倍型序列的聚类分析表明,H33(昆明)与其他单倍型存在差异(图2)。其他种群间并无明显分化。在这些确定的单倍型中,有28个是特有的。3个单倍型(H2、H13和H23)分布最广,分别占总数的8.86%、39.24%和6.33%。苏州种群有特有单倍型H29、H30、H31。H23包含苏州和武汉种群的个体。
对gnd单倍型序列的聚类分析表明,该系统发育树明显分为两大簇,其中H1和H5成一簇,其余单倍型成另一簇。泰安种群有独有单倍型H5,银川种群有特有单倍型H3和H4。单倍型H6包含昆明和银川种群个体。单倍型H1和H2包含所有种群的个体。
trpA单倍型系统发育树也分为两大簇。银川种群有最多种的稀有单倍型H13、H14、H15、H16、H17、H18。只有廊坊种群没有特有单倍型。单倍型H8包含武汉,泰安和银川种群个体。苏州种群有特有单倍型H19。单倍型H1和H2的个体分布于各个种群中。

图2 基于COⅠ (a)、gnd (b)和trpA (c)的荻草谷网蚜单倍型的邻接系统发育树Fig.2 Neighbor-joining phylogenetic trees of the haplotypes of Sitobion miscanthi from China based on COⅠ (a),gnd (b),and trpA (c)
2.4 单倍型网络图
线粒体基因COⅠ单倍型网络图显示,33个单倍型分为两组(图3a)。高频单倍型为H2、H13和H23,代表了大多数的单倍型,这3种单倍型可能是祖先单倍型。H13包含的个体数最多,有31个,分布于武汉和泰安两个种群中。H12和H13间的遗传差异较小。H2和H23分别包含7个和5个个体,其他30个单倍型包含的个体数在1~4个,有28个只包含1个个体的稀有单倍型。剩余的稀有单倍型位于网络图的边缘和连接处,并通过突变与祖先单倍型相连接。大多数稀有单倍型通过一个或多个突变与祖先单倍型相关联,少数则通过祖先单倍型的多次连续突变获得。一些单倍型缺失,可能是由于采样不足导致缺少预期的单倍型。或者由于进化,在此期间某些单倍型可能从现存的种群中消亡。泰安和武汉种群分别具有种类最多的稀有单倍型。gnd基因单倍型网络图(图3b)是一个以单倍型H1和H2为中心的小型网络图。其余的单倍型都是稀有单倍型。trpA基因单倍型形成的网络图(图3c)显示H1和H2单倍型位于网络图的中间,以这两个单倍型为中心形成了2个大型星形放射状子网络。剩余的稀有单倍型通过突变连接到H1或H2,并位于网络图的边缘。
2.5 历史种群动态
在分析种群的历史动态时,通常使用中性检验和错配分析。中性检验分析使用参数值来确定种群是稳定种群还是最近经过扩张形成的种群。理论上,在一个稳定的种群中,Fu’sFs和Tajima’sD的值接近于0;显著的负值表示种群突然扩张;显著的正值表明种群可能已经分化,导致形成亚种群,或者种群经历了瓶颈事件。在本试验中,COⅠ和trpA基因的Fu’sFs值为-1.622 74和-1.203 26,显示显著的负值,表明种群经历了扩张。
3 结论与讨论
荻草谷网蚜的主动迁飞能力有限,但可以凭借风力等多种途径传播。传统的标记回收法可用于确定大型昆虫的迁飞及迁飞途径[23],但蚜虫个体小,不易回收,因此传统的化学染料标记难以实现。

图3 基于COⅠ(a)、gnd (b) 和 trpA (c) 的中国荻草谷网蚜单倍型网络Fig.3 Haplotype network of Sitobion miscanthi from China based on COⅠ (a),gnd (b),and trpA (c)
近年来对昆虫迁飞路径的研究表明,分子生物学技术是一种分析物种扩散传播的有力手段[24]。Yang等使用COⅠ基因等标记揭示了小菜蛾Plutellaxylostella(Linnaeus)在中国从长江中下游迁飞至中国北方,最终抵达东北[25]。徐昭焕等利用COⅠ基因等标记研究了中国17个地理种群的荻草谷网蚜,推测具有不同单倍型的南方虫源会在初春迁飞至北方[10]。Prabhulinga等利用线粒体COⅠ基因对印度22个地区的棉花上的烟粉虱Bemisiatabaci(Gennadius)进行了遗传多样性和地理学结构的分析,证实了印度的烟粉虱是按地理距离隔离的[26]。同时,蚜虫的初级共生菌对寄主的分布和进化有着深远的影响,因此也有研究将初级共生菌基因作为候选基因标记。Zhang等利用蚜虫初级共生菌B.aphidicola的atpAGD基因对8个中国漆树瘿蚜Schlechtendaliachinensis不同地理种群进行了遗传多样性分析,结果表明atpAGD基因的16个单倍型被划分为被长江分隔的南北进化枝,推测当代遗传结构很可能受到历史地质事件的影响[27]。因此蚜虫初级共生菌基因可以被用来研究蚜虫不同种群间遗传多样性差异。
本研究以线粒体COⅠ基因与初级共生菌B.aphidicola的2个单拷贝基因gnd和trpA分析了6个荻草谷网蚜不同地理种群(银川、苏州、武汉、昆明、泰安、廊坊)遗传多样性和遗传结构。SAMOVA分析结果表明,6个不同地理种群可分为3组,分别为第一组银川种群,第二组苏州种群和第三组其他种群(武汉、昆明、泰安、廊坊种群),组间存在显著的遗传差异。同时,线粒体和初级共生菌基因的遗传结构分析结果几乎保持一致,表明初级共生菌在研究蚜虫种群的遗传结构和多样性方面具有潜在意义。本试验中的Fu’sFs和Tajima’sD中性检验的结果呈负值,表明我国的荻草谷网蚜种群在历史上经历了明显地种群扩张过程。
进一步的系统发育分析显示,第三组内的武汉、昆明、泰安和廊坊4个种群几乎包含了所有单倍型种类,表明这4地的种群之间存在着频繁的基因交流。而银川种群和苏州种群中亦有与这4地相同的单倍型,其中银川种群和泰安种群间有相同的稀有单倍型,苏州种群和武汉种群也有相同的稀有单倍型。3组之间存在着基因交流。我们由此推测在4、5月份正值黄淮海麦区小麦的灌浆期,且东南季风盛行的情况下,银川种群迁入了外来虫源。而在9、10月份伴随着西北季风,苏州种群也迁入了长江中下游麦区的外来虫源[28]。这与董庆周等的研究提出银川存在外来虫源,并存在远距离迁飞现象的结论一致[8]。杨素钦等在研究荻草谷网蚜的虫源地时也指出我国0℃等温线以北的山西、河北、山东大部分地区以及北京、天津等地的荻草谷网蚜可能是由河南省南部、江苏和安徽省北部及其以南地区迁入[29]。本试验的荻草谷网蚜群组划分与Wang等的研究结果[30]也基本一致。因此,相比于传统的研究昆虫迁飞的方法,利用分子标记可以更加方便和快速地研究昆虫种群遗传多样性及推测扩散路径。此外,本研究表明初级共生菌Buchnera的两个单拷贝基因gnd与trpA可以作为研究蚜虫种群遗传多样性和遗传结构的分子标记。进一步证明了昆虫共生菌相关基因可作为新型分子标记,并且可与传统的线粒体COⅠ基因等在研究中相互佐证。
综上所述,本文基于线粒体基因和共生菌基因,探明了中国小麦主产区荻草谷网蚜6个不同地理种群遗传多样性,研究结果为麦蚜迁飞途径及虫源关系的预测提供了分子生态学技术手段,具有较强的理论意义。
