中国4省猕猴桃细菌性溃疡病菌的生物型检测和遗传多样性分析
2022-12-19代玉立兰成忠刘晓菲龚国淑杨秀娟
代玉立,兰成忠,甘 林,刘晓菲,龚国淑,杨秀娟*
(1.福建省农业科学院植物保护研究所,福建省作物有害生物监测与治理重点实验室,福建省作物有害生物绿色防控工程研究中心,福州 350013;2.四川农业大学农学院,成都 611130)
猕猴桃ActinidiachinensisPlanch.是我国重要的经济水果,其种植面积和产量均位于世界前列[1]。由丁香假单胞菌猕猴桃致病变种Pseudomonassyringaepv.actinidiae(Psa)引起的猕猴桃细菌性溃疡病(kiwifruit bacterial canker)是全球猕猴桃产业最具毁灭性的病害,该病害暴发速度快、毁灭性极强且难以防治[1-4]。猕猴桃细菌性溃疡病最早发生于日本[5],此后在韩国[6]、意大利[7]、新西兰[8]、西班牙[9]和法国[10]等猕猴桃生产国流行暴发,给猕猴桃安全生产造成了极大的威胁。我国于1985年首次在湖南省发现此病[11],1989年该病害在四川省造成猕猴桃植株大面积死亡[12],1991年该病害在安徽省岳西县暴发,果园株发病率达32%,造成严重经济损失[13],2018年猕猴桃细菌性溃疡病又在福建省福安市暴发,果园株发病率达70%以上,造成超过15 hm2果园被毁[14]。随着我国猕猴桃栽培的产业化和种植面积的规模化,细菌性溃疡病带来的威胁日益严峻。
明确病原菌的生物型(biovar)和群体遗传关系,有助于揭示该病原菌的适生性、起源和流行趋势[15-16]。近年来,国外研究者通过DNA指纹图谱和基因序列分析将猕猴桃细菌性溃疡病菌分成6个不同的生物型,分别命名为biovar 1[17]、biovar 2[17]、biovar 3[18]、biovar 4[18-19]、biovar 5[20]和biovar 6[21]。biovar 1主要在日本以及意大利2008年以前的猕猴桃细菌性溃疡病菌群体中发现,能产生菜豆毒素(phaseolotoxin);biovar 2仅在韩国流行危害,能产生冠菌素(coronatine),但是不产生菜豆毒素;biovar 3是分布最广、危害最严重的世界性流行群体,既不产生菜豆毒素,也不产生冠菌素;biovar 4最早发现于新西兰和澳大利亚,为弱致病力群体,仅在叶片上引起少许坏死斑,不引起溃疡和幼芽枯死;biovar 5和biovar 6是近期发现的新类型,目前仅分布于日本局部地区[1,18-19]。biovar 3因其流行范围广、适生性强和极具破坏性而引起国内外学者的研究兴趣[1,3,15,22]。目前,我国研究者对猕猴桃细菌性溃疡病的研究主要在病原物的分子检测与鉴定、病害流行规律及防控等方面[23],而有关我国猕猴桃细菌性溃疡病菌的生物型检测研究相对较少。随着植物病原菌基因组测序和核苷酸组成结构研究的深入,分子标记技术被广泛应用于植物病原菌遗传多样性和遗传结构的研究中,如随机扩增多态性(RAPD)、扩增片段长度多态性(AFLP)、限制性内切酶片段长度多态性(RFLP)、微卫星分子标记(microsatellite)、单核苷酸多态性(SNP)、短重复序列标记(rep-PCR)等[1,23-24]。多基因联合分析技术可从DNA分子组成上对单个碱基差异进行检测,揭示种群内不同个体间的遗传多样性,被广泛应用于植物病原细菌群体分型和分子进化关系研究中。研究表明,植物病害暴发成灾与病原菌群体演替和遗传多样性及遗传结构改变关系密切[23]。因此,了解病原菌群体在分子水平上的遗传多样性,不仅可以揭示病害暴发成灾的内在机制,还可为病害的长期可持续控制提供理论指导。
前期基于部分省份的猕猴桃细菌性溃疡病菌群体遗传多样性研究表明,猕猴桃细菌性溃疡病菌存在丰富的DNA多态性,且其DNA多态性与地理来源和寄主品种无明显相关性,造成这种遗传差异的原因以及我国不同地理来源的猕猴桃细菌性溃疡病菌种群间的遗传关系未见深入研究。因此,笔者拟采用检测猕猴桃细菌性溃疡病菌生物型的特异性引物,测定福建、安徽、四川和陕西4省猕猴桃细菌性溃疡病菌群体的生物型。采用多基因联合分析4省猕猴桃细菌性溃疡病菌的遗传多样性,以期确定福建省猕猴桃细菌性溃疡病菌的来源,为阻断猕猴桃细菌性溃疡病菌在福建省的传播提供理论参考。
1 材料与方法
1.1 供试菌株
供试的安徽省猕猴桃细菌性溃疡病菌菌株由安徽农业大学植物保护学院张立新教授惠赠;四川省猕猴桃细菌性溃疡病菌菌株由四川农业大学农学院龚国淑教授惠赠;福建和陕西省猕猴桃细菌性溃疡病菌菌株由福建省农业科学院植物保护研究所植物病理研究室分离保存。猕猴桃细菌性溃疡病菌菌株的编号、菌株数、采集年份和来源见表1。

表1 供试的4省猕猴桃细菌性溃疡病菌信息Table 1 Information of tested strains of Pseudomonas syringae pv.actinidiae from four provinces
1.2 供试菌株基因组DNA提取
将用30%甘油保存在-80℃超低温冰箱中的猕猴桃细菌性溃疡病菌菌株于28℃培养箱中孵育30 min。取1 mL菌悬液接种到100 mL液体LB培养基中,28℃振荡(180 r/min)培养活化24 h后,取1 mL菌悬液接种到100 mL LB培养基中,相同条件下振荡培养24~48 h。取2 mL菌悬液于2 mL无菌离心管中,10 000 r/min离心2 min,弃上清,收集细胞。猕猴桃细菌性溃疡病菌的基因组DNA提取采用ONE-4-ALL基因组DNA小量提取试剂盒[生工生物工程(上海)股份有限公司],操作步骤严格按试剂盒说明书进行。DNA样品用50 μL TE缓冲液(10 mmol/L Tris-HCl,1 mmol/L EDTA,pH 8.0)溶解,核酸定量仪测定各菌株DNA浓度并用TE缓冲液调节至100 ng/μL,DNA样品保存于-20℃冰箱。
1.3 猕猴桃细菌性溃疡病菌的生物型检测
猕猴桃细菌性溃疡病菌biovar 1、biovar 2、biovar 3、biovar 5和biovar 6等5种生物型检测采用Lee等[25]、Koh等[26]以及Fujikawa等[20]设计的生物型检测特异引物(表2)。引物委托生工生物工程(上海)股份有限公司合成。由于biovar 4未被学术界广泛认可,故本文未作首选检测,若排除前5种生物型,则考虑检测biovar 4。
生物型检测PCR扩增体系(25 μL):2×PremixTaqMasterMix 12.5 μL(大连宝生物有限公司),引物各10 pmol,DNA模板100 ng,补足ddH2O至25 μL;PCR扩增条件:94℃预变性5 min;94℃变性45 s,最适退火温度(表2)退火45 s,72℃延伸45 s,35个循环;72℃延伸10 min。PCR扩增在C1000TM型热循环仪(美国BIO-RAD公司)中进行。PCR结束后取5 μL扩增产物于120 V恒定电压下用含EB的1.2%琼脂糖凝胶电泳20 min,BioDoc-ItTM型凝胶成像系统(美国UVP公司)检测并拍照。

表2 供试引物信息1)Table 2 Information of tested primers
1.4 猕猴桃细菌性溃疡病菌群体的遗传多样性分析
1.4.1基因序列特征和单倍型
本研究采用16S rRNA、ITS、gapA和rpoD等4个基因用于猕猴桃细菌性溃疡病菌遗传多样性分析。引物详细信息见表2。引物委托生工生物工程(上海)股份有限公司合成。25 μL PCR扩增体系:2×PremixTaqMasterMix 12.5 μL(大连宝生物有限公司),引物10 pmol,DNA模板100 ng,补足ddH2O至25 μL;PCR扩增条件:94℃预变性5 min;94℃变性45 s,最适退火温度(表2)退火45 s,72℃延伸45 s(16S rRNA延伸90 s),35个循环;最后72℃延伸10 min。PCR结束后取25 μL扩增产物于120 V恒定电压下用含EB的1.5%琼脂糖凝胶电泳25 min,BioDoc-ItTM型凝胶成像系统检测并拍照。紫外灯下用无菌手术刀切取含目的条带的琼脂糖凝胶,采用凝胶回收试剂盒(AXYGEN公司)回收。目的基因由生工生物工程(上海)股份有限公司测序。
测序产物利用DNAMAN软件分别进行单基因多重比对,ClustalX软件[32]进行对齐,再利用DnaSP 6软件[33]将4个单基因序列串联,分析串联序列整体特征:G+C含量、总突变位点数、多态性位点数(number of polymorphic sites;Ns)、核苷酸多样性(nucleotide diversity;Pi)、平均核苷酸差异数(average number of nucleotide differences;K)、单倍型(haplotypes)以及单倍型多样性(haplotype diversity;Hd)。
1.4.2猕猴桃细菌性溃疡病菌群体遗传多样性
采用DnaSP 6软件分析中国猕猴桃细菌性溃疡病菌不同地理来源群体间的单倍型数、单倍型多样性、多态性位点数、核苷酸多样性以及平均核苷酸差异数等遗传多样性参数,比较不同群体间的遗传多样性,采用Tajima’sD和Fu’sF对4个猕猴桃细菌性溃疡病菌群体进行中性检验。
1.4.3猕猴桃细菌性溃疡病菌群体遗传分化
将多基因串联序列输入DnaSP 6软件分析中国猕猴桃细菌性溃疡病菌不同地理来源群体间的遗传分化系数Fst和基因流Nm[Nm=0.25(1-Fst)/Fst],当Fst≤0.05:遗传分化水平极低,0.05
1.4.4猕猴桃细菌性溃疡病菌群体遗传距离和聚类分析
杨梅(Myrica rubra Sieb. et Zucc)原产于我国南方,主要分布在浙江、江苏、江西、福建等省,其中以浙江省所产杨梅品质最佳。杨梅果实色泽艳丽、酸甜多汁、营养丰富,深受消费者的欢迎。杨梅果实除含有多种人体必需的营养物质(如糖类、维生素和矿质元素)之外,还含有大量的花色苷和多酚类物质,因此具有较好的自由基清除能力。此外杨梅还具有生津消渴、解酒解暑、行气止痛等保健功效,被人们赞誉为“初夏江南珍果”。
利用MEGA 7软件[35]分析猕猴桃细菌性溃疡病菌不同菌株和不同地理来源群体间的遗传距离,利用遗传距离数据和GenAlEx 6.5软件中主坐标分析(principal co-ordinates analysis,PCoA)分析猕猴桃细菌性溃疡病菌不同地理来源群体内不同个体间的遗传差异。利用遗传距离数据和MEGA 7软件采用邻近法(neighbor-joining)构建系统发育树。
2 结果与分析
2.1 猕猴桃细菌性溃疡病菌的生物型
通过biovar 1、biovar 2、biovar 3、biovar 5和biovar 6共5对猕猴桃细菌性溃疡病菌生物型检测特异性引物对我国4个地理种群的47株菌株进行PCR扩增,仅biovar 3特异性引物可以扩增出一条545 bp的特异性条带(图1),表明供试菌株的生物型均为biovar 3。
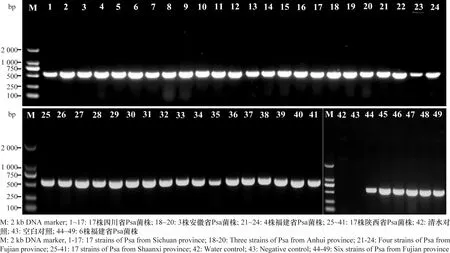
图1 中国4省猕猴桃细菌性溃疡病菌的生物型检测Fig.1 Detection of Pseudomonas syringae pv.actinidiae biovars from four provinces in China
2.2 猕猴桃细菌性溃疡病菌群体遗传多样性分析
2.2.1基因序列特征和单倍型
通过对我国猕猴桃细菌性溃疡病菌的16S rRNA、ITS、gapA和rpoD基因序列进行联合,获得一条全长2 743 bp的同源序列。联合基因G+C含量为57.3%,总突变位点数为95,多态性位点数为87,核苷酸多态性为0.002 21,平均核苷酸差异数为5.880。
通过对我国4省猕猴桃细菌性溃疡病菌16S rRNA、ITS、gapA和rpoD基因进行联合分析,从47个菌株中共检测出27个单倍型(Hap01~Hap27),单倍型多样性为0.955。Hap01为安徽省猕猴桃细菌性溃疡病菌群体特有单倍型;Hap02、Hap10、Hap11、Hap13和Hap16为群体共有的单倍型,其中Hap02共享范围最广,为福建、四川和陕西群体共有;Hap03、Hap04、Hap05、Hap06、Hap07、Hap08和Hap09为福建省猕猴桃细菌性溃疡病菌群体特有单倍型;Hap12、Hap14、Hap15、Hap17、Hap18、Hap19和Hap20为四川省猕猴桃细菌性溃疡病菌群体特有单倍型;Hap21、Hap22、Hap23、Hap24、Hap25、Hap26和Hap27为陕西省猕猴桃细菌性溃疡病菌群体特有单倍型(表3)。

表3 中国4省猕猴桃细菌性溃疡病菌的单倍型分布Table 3 Haplotype distributions of Pseudomonas syringae pv.actinidiae populations from four provinces in China
2.2.2猕猴桃细菌性溃疡病菌群体遗传多样性
来自我国4省猕猴桃细菌性溃疡病菌群体的单倍型数(Nh)差异较大,安徽省群体仅有1个单倍型,而四川和陕西群体单倍型数达12个;单倍型多样性(Hd)介于0~0.971之间。4个群体间的多态性位点数(NS)差异极显著(P<0.01),其中安徽群体的NS最低,福建群体的NS最丰富;4个群体的核苷酸多样性(Pi)和平均核苷酸差异数(K)范围分别为0~0.005 69和0~15.356,也是安徽和福建群体分别为最低和最高;除安徽群体外,其他3个群体的Tajima’sD和Fu’sF均小于0,表明3个群体中稀有等位基因以高频率存在,是非平衡选择的结果(表4)。
2.2.3猕猴桃细菌性溃疡病菌群体遗传分化
AMOVA分析表明,3.6%的遗传变异来源于群体间,而96.4%的遗传变异来源于群体内,说明种群内变异是遗传变异的主要来源(表5)。遗传分化指数(Fst)和基因流(Nm)分析结果表明,安徽省猕猴桃细菌性溃疡病菌群体与福建、四川和陕西群体间存在中等到极高的遗传分化,Fst值介于0.175~0.428之间(Fst>0.15),Nm值较小(0.334~1.179),说明群体间缺乏基因交流。福建、四川和陕西两两群体间的遗传分化水平均较低,Fst值介于-0.015~0.017之间,Nm值较大(>14),说明群体间基因交流频繁(表6)。AMOVA分析也表明,群体间的遗传分化水平极低(Fst=0.036,P<0.05)(表5)。
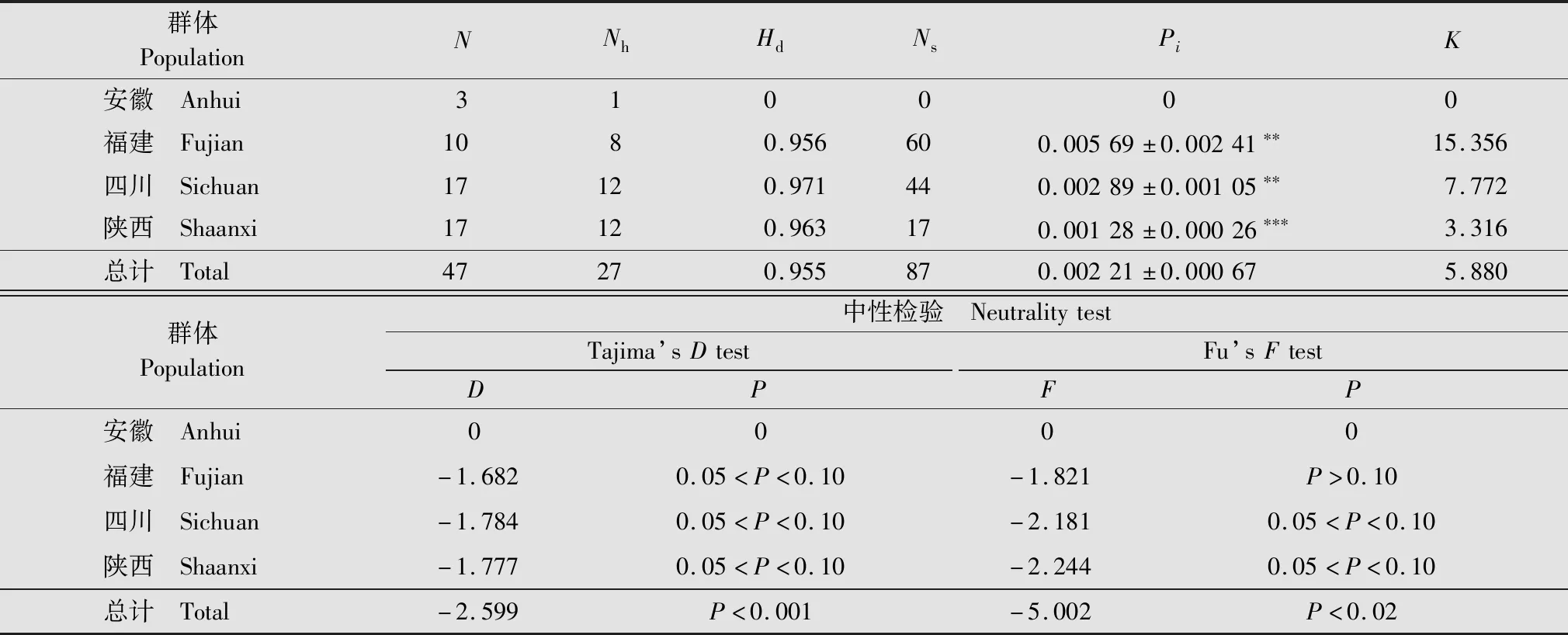
表4 中国4省猕猴桃细菌性溃疡病菌群体的遗传多样性1)Table 4 Genetic diversity of Pseudomonas syringae pv.actinidiae populations from four provinces in China

表5 中国4省猕猴桃细菌性溃疡病菌种群内和种群间的分子变异分析Table 5 Analysis of molecular variance (AMOVA) of genetic variation within and among four populations of Pseudomonas syringae pv.actinidiae in China
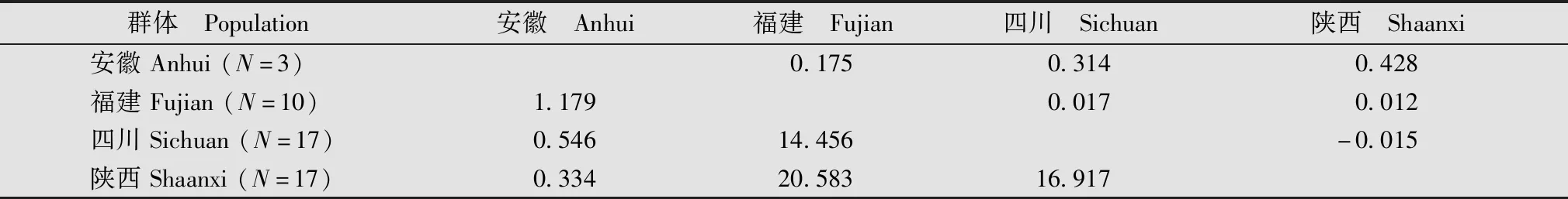
表6 中国4省猕猴桃细菌性溃疡病菌群体间的遗传分化和基因流1)Table 6 Pairwise matrix of genetic differentiation and gene flow among Pseudomonas syringae pv.actinidiae populations from four provinces in China
2.2.4猕猴桃细菌性溃疡病菌群体遗传距离和聚类分析
利用猕猴桃细菌性溃疡病菌16S rRNA、ITS、gapA和rpoD的联合基因序列,采用MEGA7软件计算4个群体间的遗传距离,结果表明:安徽、福建、四川和陕西4个猕猴桃细菌性溃疡病菌群体间的遗传距离为0.001 45~0.004 64,其中安徽与陕西群体间的遗传距离最近,而福建与安徽、四川和陕西群体间的遗传距离均较远(表7)。PCoA分析结果表明,第一和第二主坐标分别占总变异的67.16%和28.40%,4个不同地理来源的猕猴桃细菌性溃疡病菌个体之间没有明显的分离(图2)。基于不同个体间遗传距离的聚类分析结果表明,除安徽群体外,不同地理来源的猕猴桃细菌性溃疡病菌个体随机聚于各个分支中,说明3个地理来源的猕猴桃细菌性溃疡病菌的亲缘关系较近(图3)。
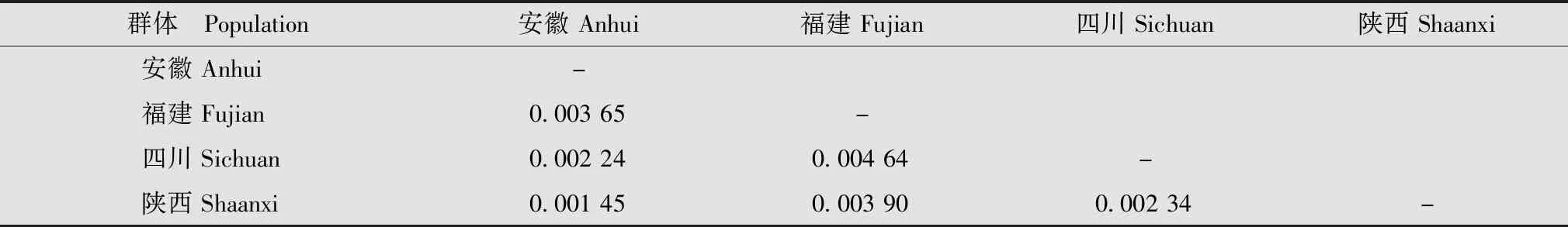
表7 中国4省猕猴桃细菌性溃疡病菌群体间的遗传距离Table 7 Genetic distance between different populations of Pseudomonas syringae pv.actinidiae from four provinces in China

图2 中国4省猕猴桃细菌性溃疡病菌群体的PCoA分析Fig.2 Principal co-ordinates analysis (PCoA) of Pseudomonas syringae pv.actinidiae populations from four provinces in China
3 讨论
Vanneste等[18]根据致病性、表型和基因组特征,将来自日本和意大利(1992年)的猕猴桃细菌性溃疡病菌群体的生物型定义为biovar 1,来自韩国的猕猴桃细菌性溃疡病菌群体定义为biovar 2,来源于中国的猕猴桃细菌性溃疡病菌群体定义为biovar 3。Cunty等[36]通过可变数目重复序列分析(multilocus variable-number tandem-repeat analysis,MLVA)发现,来自中国5省的猕猴桃细菌性溃疡病菌菌株和来自法国、意大利以及新西兰的菌株聚于同一分支,尽管这些菌株的来源地不同,但它们都是同一生物型biovar 3。本研究通过生物型特异性引物检测发现,供试的4省47株猕猴桃细菌性溃疡病菌菌株的生物型均为biovar 3,未发现其他生物型,研究结果与前人报道一致。鉴于当前在我国未发现其他猕猴桃细菌性溃疡病菌生物型,因此,应加强检疫措施,防止外来生物型菌株的入侵,保障我国猕猴桃产业的健康可持续发展。
对分离自安徽、福建、四川和陕西4省的47株猕猴桃细菌性溃疡病菌的16S rRNA、ITS、gapA和rpoD基因序列进行联合分析,共获得2 743 bp的有效同源序列,联合基因中G+C含量为57.3%,与任雪燕等[23]基于我国7省的21个猕猴桃细菌性溃疡病菌菌株全基因组测序获得的G+C含量相当,略低于丁香假单胞菌丁香致病变种P.syringaepv.syringae的全基因组G+C含量[37]。来自4省47株猕猴桃细菌性溃疡病菌群体共检测到27个单倍型,其中安徽群体的单倍型数最少,仅有1种单倍型;福建、四川和陕西群体的单倍型数相当,分别为8、12个和12个,单倍型多样性指数分别为0.956、0.971和0.963。Sawada等[38]通过致病性相关基因分析表明,来自全球范围的猕猴桃细菌性溃疡病菌不同生物型间存在显著的遗传多样性,且这种多样性与致病力、基因型和地理来源有关。Ferrante等[39]通过rep-PCR指纹图谱研究全球早期和现阶段猕猴桃细菌性溃疡病菌流行群体的遗传结构表明,全球猕猴桃细菌性溃疡病菌存在丰富的遗传多样性。He等[40]通过rep-PCR、RAPD和IS50-PCR等3种分子标记技术,发现我国5个地理种群的biovar 3型猕猴桃细菌性溃疡病菌存在丰富的遗传多样性。任雪燕等[23]通过全基因组测序研究我国7省21株猕猴桃细菌性溃疡病菌的遗传多样性,表明不同地理群体间的核苷酸多态性差异不明显。本研究结果表明,来自我国4省的猕猴桃细菌性溃疡病菌存在较丰富的遗传多样性,基于16S rRNA、ITS、gapA和rpoD基因序列的总突变位点数为95,多态性位点数为87,核苷酸多态性为0.002 21,平均核苷酸差异数为5.880。来自福建群体的遗传多样性最丰富,其次为四川群体,安徽群体的遗传多样性最差,4个群体间的核苷酸多态性存在极显著差异(P<0.01)。本研究结果与Sawada等[38],Ferrante等[39]以及He等[40]研究结果一致,而与任雪燕等[23]研究结果不吻合,原因可能是分析方法和角度不同造成的[23]。
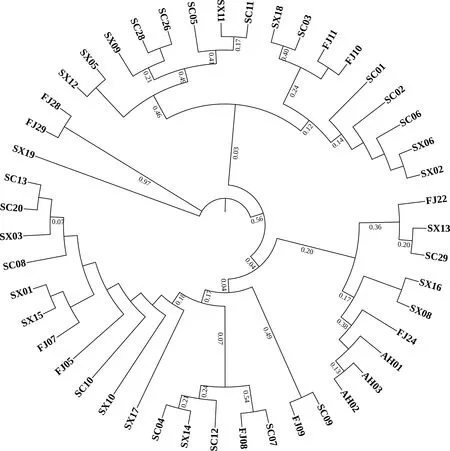
图3 中国4省猕猴桃细菌性溃疡病菌的聚类分析Fig.3 Phylogenetic analysis of Pseudomonas syringae pv.actinidiae from four provinces in China
遗传分化指数和基因流是衡量遗传分化水平的2个重要指标,遗传分化指数越大,基因流越小,说明群体间基因交流越匮乏,遗传分化水平高[23,41]。安徽群体与福建、四川和陕西群体病原菌之间的基因流均较小,遗传分化十分明显(Fst介于0.175~0.428之间),究其原因可能是安徽省是我国猕猴桃细菌性溃疡病菌原始发现地之一,与其他3个群体来源不同[13]。福建群体病原菌与四川和陕西2个群体病原菌的基因交流十分频繁(Nm>14),从而导致了极低水平的遗传分化(Fst介于-0.015~0.017之间),主要原因可能是福建省猕猴桃细菌性溃疡病菌是外来入侵种。近年来福建省种植的猕猴桃品种大多为‘红阳’品种,而‘红阳’猕猴桃是由四川省苍溪县繁育而成[42],此外,陕西和四川省还是我国重要的猕猴桃苗木培育基地,随着猕猴桃苗木输送到全国各地,造成病原菌随苗木传播[23]。因此,应强化猕猴桃苗木繁育基地的检疫措施,阻断病原菌随苗木调运的传播。
PCoA分析和聚类分析表明,除安徽病原菌群体外,来自福建、四川和陕西省的猕猴桃细菌性溃疡病菌较为分散,没有表现出较强的地理来源相关性。但是,本研究结果不能得出我国猕猴桃细菌性溃疡病菌的遗传多样性与地理来源无关的结论,主要原因在于福建省的菌源为外来菌株,可能随苗木的调运从四川和陕西省传入福建。安徽群体表现出较强的地理相关性,但是由于样本数量有限,说服力不强。由于我国猕猴桃细菌性溃疡病菌的来源地较多(如湖南、安徽和四川等)[11-13],因此,作者推测我国猕猴桃细菌性溃疡病菌的遗传多样性可能与该菌的地理来源关系较为密切。有关我国猕猴桃细菌性溃疡病菌的遗传多样性与地理来源关系密切的推论是否成立,仍需进一步扩大采样的时间和空间范围,作进一步深入探讨。
4 结论
利用猕猴桃细菌性溃疡病菌生物型PCR检测特异性引物,明确了来自安徽、福建、四川和陕西4省的猕猴桃细菌性溃疡病菌的生物型均为biovar 3。采用猕猴桃细菌性溃疡病菌的16S rRNA、ITS、gapA和rpoD基因联合分析方法,从DNA分子水平上揭示了安徽、福建、四川和陕西4省的猕猴桃细菌性溃疡病菌群体间存在遗传多样性,且不同病原菌群体的遗传多样性差异不同。研究结果为明确福建省猕猴桃细菌性溃疡病菌的来源、阻断其在福建省的传播以及该病害的长期可持续控制提供了理论参考。
