鱼露中氯丙醇快速筛查方法的探讨
2022-12-17蒋林惠袁琛凯丁红梅
蒋林惠,石 敏,袁琛凯,丁红梅
南通市食品药品监督检验中心 (南通 226006)
鱼露,别名鱼酱油,是一种以低值鱼虾等水产品为原料,经腌渍、发酵、熬制等工艺所得的液体调味料。鱼露原产于我国东南沿海地带,其酿造历史已达两千多年,是我国传统发酵调味品的精粹。因其独特的风味与营养价值使得鱼露逐步受到世界各地居民的喜爱,被广泛应用于食品加工业及餐饮行业[1,2]。氯丙醇类化合物是食品及食品接触材料中的一类污染物,其对人体健康有着潜在的伤害,国际上的专家学者经研究发现,氯丙醇类化合物具有生殖、肾脏、神经毒性,并且有致癌、致突变及诱导细胞凋亡的风险[3-6]。
食品及接触材料的氯丙醇污染物来源较多,但鱼露类调味品类的氯丙醇污染物来源主要为水解植物蛋白液。一些生产企业为缩短调味品的发酵周期,获得最大经济利益,过量采用水解植物蛋白液,最终易导致其氯丙醇类化合物超过标准规定限量值[7,8]。GB 5009.191—2016《食品安全国家标准 食品中氯丙醇及其脂肪酸酯含量的测定》中第二法中氯丙醇类化合物检出限为3-MCPD、2-MCPD为2 μg/kg,1,3-DCP、2,3-DCP为5 μg/kg。液态调味品中加了酸水解蛋白液的3-MCPD限量值为0.4 mg/kg。这两者都给实验检测的精密度、准确度提出了不小的挑战。
按照已有的国标方法进行氯丙醇类化合物的检测,其主要方法是将氯丙醇经衍生后再采用气相色谱—质谱进行单离子扫描分析,间接对氯丙醇进行定量[9-11]。但鱼露基质复杂,衍生化实验后易产生多种干扰物质,会污染色谱柱并将影响目标化合物的定性定量[12]。为改善上述问题,将气质联用的多反应监测模式与同位素内标法定量结合,采用QuECHERS前处理技术进行目标化合物纯化,该方案无需衍生化,可直接测定氯丙醇类化合物,更加高效、集约、准确。
1 材料与方法
1.1 仪器和试剂
MIKRO 220R型台式高速冷冻离心机,德国Hettich科学仪器公司;Agilent 7890B/7000D型安捷伦气相色谱三重四级杆质谱联用仪,美国安捷伦科技公司;VM-2500型,多管旋涡混匀仪,广东安胜仪器有限公司;BSA 224S型电子天平,赛多利斯科学仪器(北京)有限公司;DB-WAX UI型气相色谱柱(30 m×0.25 mm,0.25 μm),美国安捷伦科技公司。
D5-1,3-二氯-2-丙醇、3-氯-1,2-丙二醇、2,3-二氯-1-丙醇、1,3-二氯-2-丙醇,阿尔塔(天津)标准物质研究院有限公司;D5-2,3-二氯-1-丙醇、D5-3-氯-1,2-丙二醇,上海安谱璀世标准技术服务有限公司;D5-2-氯-1,3-丙二醇、2-氯-1,3-丙二醇,北京曼哈格生物科技有限公司;甲醇、正己烷、乙酸乙酯,色谱纯,德国默克公司;无水硫酸镁、氯化钠,优级纯,国药集团化学试剂有限公司;C18(十八烷基硅烷)、PSA(N-丙基乙二胺),优级纯,阿拉丁试剂(上海)有限公司;有机相针式滤头,0.22 μm,上海安谱实验科技股份有限公司。
1.2 方法
1.2.1标准工作曲线的配置
选择氯丙醇类化合物及同位素内标的标准储备液(100 mg/L),本实验配制线性范围1~1 000 μg/L的标准曲线(其对应内标物质含量为100 μg/L,试剂为乙酸乙酯),标准系列工作液直接进入GC-MS-MS分析。
1.2.2样品前处理方法
实验选择鱼露作为实验样品。准确称取样品2.0 g于50 mL塑料离心管中,加入混合内标溶液,超声5 min,涡旋混匀后待处理。再加入2 mL甲醇、QuECHERS试剂包(无水硫酸镁2 g,氯化钠0.5 g,C18 100 mg,PSA 100 mg),涡旋5 min,10 000 r/min离心,上清液转移至5 mL容量瓶,重复提取两次,合并上清液并定容至5 mL。取上清液过0.22 μm有机针式滤头后进行GC-MS-MS测定。
1.2.3气相色谱分析参数
色谱柱为DB-WAX。程序升温:初始温度50 ℃,以10 ℃/min升至180 ℃,保持5 min;再以30 ℃/min升至230 ℃,保持5 min;后运行为310 ℃,2 min。进样体积1 μL,不分流进样。碰撞气为高纯氮气,流速1.5 mL/min。载气为高纯氦气,流速2.25 mL/min。
1.2.4串联质谱分析参数[13]
离子源:电子轰击源(EI)70 eV,温度230 ℃。溶剂延迟3 min。碰撞气为高纯氮气,流速1.5 mL/min。采用多反应监测模式(dMRM)扫描,主要质谱参数见表1。
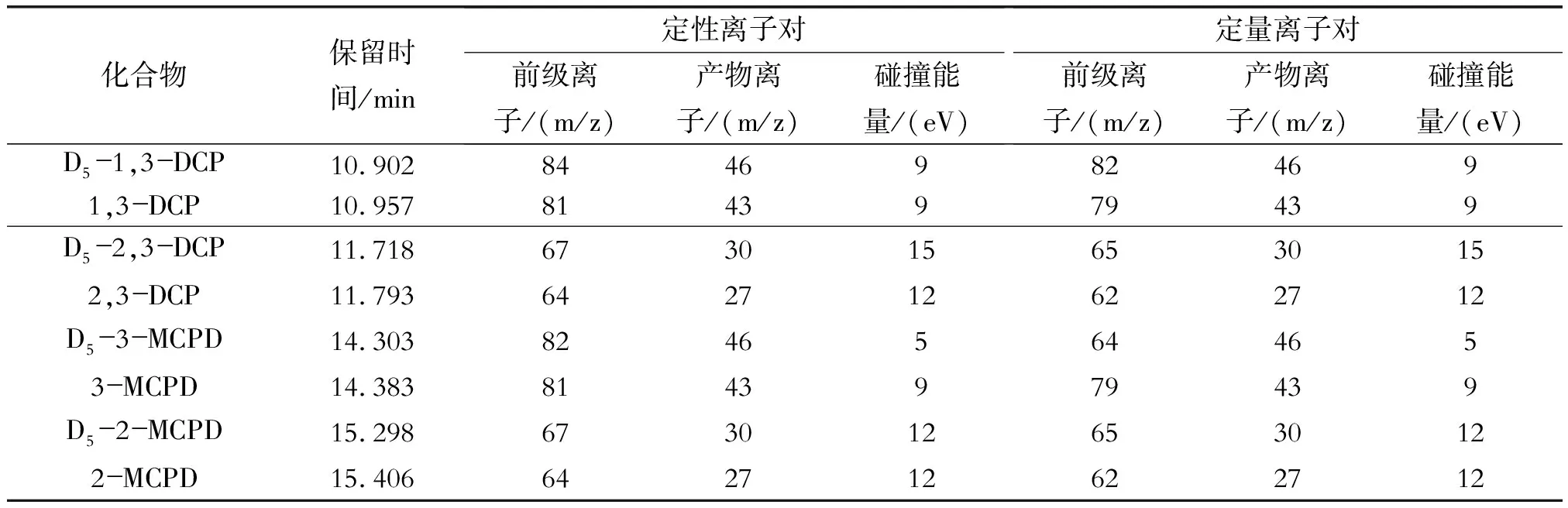
表1 氯丙醇类化合物的主要质谱信息
1.3 数据分析
目标化合物氯丙醇的含量用同位素内标法定量。采用安捷伦MS Hunter工作站进行数据采集与处理,SPSS 21.0进行数据整理分析。
2 结果与讨论
2.1 样品前处理方法的优化
氯丙醇类化合物极易溶于甲醇、乙腈和水等极性溶液,将这三者进行试验比较,最终选择甲醇作为提取试剂。甲醇能将鱼露中的一些物质进行沉淀,并且该溶剂可直接进入气质联用仪分析,避免溶剂替换。无水硫酸镁与氯化钠四比一的经典配比能够产生有效盐析,另外C18和PSA能吸附鱼露中的多种杂质,包括甾醇、有机酸、脂肪酸等[14,15]。实验中选择甲醇-固相分散萃取试剂进行液态调味品的快速前处理,所得上清液能够直接进行GC-MS-MS分析,并且其最终回收率较高,详见表2。

表2 空白鱼露样品加标回收实验结果(n=6)
2.2 色谱柱的选择
氯丙醇类物质均属强极性化合物。若直接进行气相色谱串联质谱分析则需要选择极性色谱柱。本实验选择DB-WAX强极性色谱柱进行分析,结果峰型好,分离效果优,详见图1。
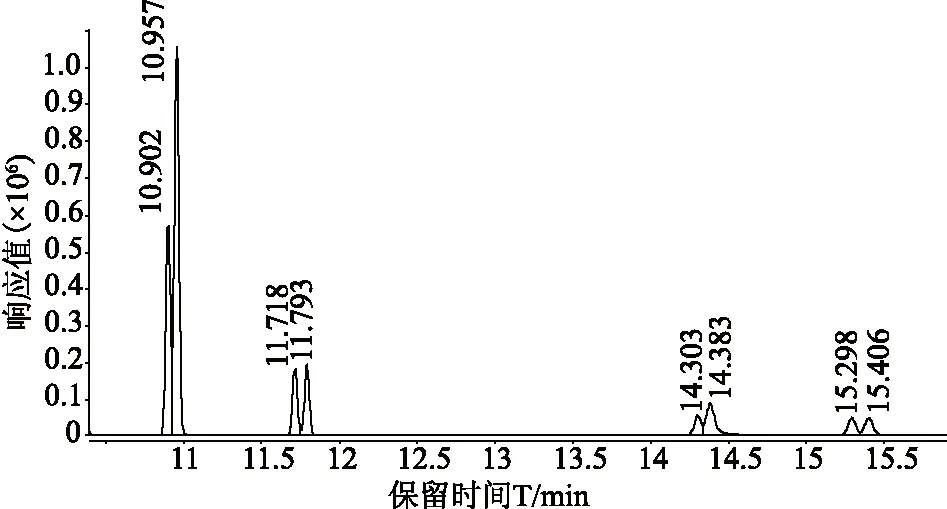
注:保留时间对应化合物分别为10.902(D5-1,3-DCP)、10.957(1,3-DCP)、11.718(D5-2,3-DCP)、11.793(2,3-DCP)、14.303(D5-3-MCPD)、14.383(3-MCPD)、15.298(D5-2-MCPD)、15.406(2-MCPD)。 图1 氯丙醇及同位素内标物质的混合标准溶液(1 mg/L)总离子流图
2.3 质谱条件的优化
基于氯丙醇类化合物的标准溶液及内标工作液,实验选择多反应监测模式进行目标化合物的数据采集分析,通过前级离子、产物离子及多反应监测模式下的碰撞能量优化。首先进行一级质谱扫描获得目标化合物的保留时间及主要碎片离子,选择离子丰度最高的2个碎片离子作为前级离子。基于前级离子设置碰撞能量9 V,采集质荷比(m/z)20~90范围内的碎片离子。结果发现8种目标化合物均仅有一个碎片离子的响应值突出,故仅选用一个产物离子。在确定前级离子及产物离子后进行碰撞能量的优化,设置四阶梯(5 V,9 V,12 V,15 V)的碰撞能量,考察其对应能量下离子对响应值,选择最高离子响应值的碰撞能量最为最优能量。氯丙醇类化合物的MRM质谱图如图2,各化合物的前级离子、产物离子及最优碰撞能量见表1。

图2 氯丙醇类化合物及同位素内标物质的MRM质谱图
2.4 标准曲线及检出限(基质效应)
基于氯丙醇类物质标准储备液,配制线性浓度1、5、10、20、40、100、200、400、1 000 μg/L的标准曲线(其对应内标物质含量为100 μg/L,试剂为乙酸乙酯),直接进样GC-MS-MS分析。以氯丙醇与其内标物的相对响应作为纵坐标(Y),相对浓度作为横坐标(X),绘制标准工作曲线,标准曲线详细信息详见表3。4种氯丙醇在0.001~1.0 mg/L范围内均能够呈现良好的线性关系,相关系数(R)均达0.999。本实验的检出限与国家标准GB 5009.191—2016中第二法的检出限相当,能够满足日常检测需求。
鱼露基质较为复杂,采用QuECHERS净化包,甲醇提取后能够有效的降低基质效应对于目标化合物在定性定量方面的干扰,为进一步降低基质效应,采用同位素内标做加标进行内标法定量确保检测的准确性。

表3 氯丙醇的线性方程及检出限
2.5 精密度和加标回收实验
以空白鱼露作为样本进行加标回收率和方法精密度实验,将相对标准偏差(RSD)表示方法的精密度。空白鱼露样品中加入1,3-DCP、2,3-DCP、3-MCPD、2-MCPD的标准品及对应内标,进行3水平6平行实验。加标浓度分别为10 μg/kg、100 μg/kg、500 μg/kg,结果详见表2。经统计发现,4种氯丙醇类化合物3个加标水平的回收率和相对标准偏差(RSD)分别为1,3-DCP:65.9%~97.4%(1.86%~1.03%);2,3-DCP:80.7%~98.5%(1.71%~0.74%);3-MCPD:81.6%~90.2%(8.25%~4.77%);2-MCPD:82.8%~105.3%(9.55%~1.97%)。结果表明,经本方法测定所得的方法回收率和精密度均能购满足日常检测需求。空白样品加标实验也能验证该实验方案能够有效的降低基质效应对于氯丙醇结果的准确度影响。
3 结论
本实验基于QuECHERS前处理技术结合气相色谱串联质谱仪,进行鱼露中氯丙醇的快速测定。通过对检出限、线性方程、空白样品加标回收率、加标精密度等方法学实验验证。通过验证确认,本实验方法能够满足目前对于鱼露中氯丙醇的检测要求。与国标方法相比,本方法无需衍生化反应,更加简便、高效、集约。
