草铵膦母药及水剂的高效液相色谱分析方法
2022-12-08杜秀利王广成许艳秋
杜秀利,王广成,许艳秋
[1.泸定县农产品质量安全(质量检测)中心,四川 泸定 626100;2.四川省农药检定所,成都 610041]
草铵膦(glufosinate-ammonium),化学名称4-[羟基(甲基)膦酰基]-D,L-高丙氨酸,最早于20 世纪80 年代由德国赫斯特公司开发,是一种广谱、高效、低毒的有机磷类灭生性除草剂。与传统除草剂百草枯和草甘膦相比,草铵膦具有更优良的性能[1]。近年来由于我国百草枯的禁用和抗草铵膦转基因作物及多抗转基因作物的推广应用等原因,草铵膦成为全球市场增长最快的非选择性除草剂,而中国是草铵膦的生产大国和出口大国[2]。
目前我国登记的草铵膦原药产品有60 个,母药产品有9 个。由于生产加工工艺和技术指标的要求不同,在生产过程中对母药中外来物质和杂质要求不如原药高,母药中可能含有少量添加剂和稀释剂[3],使得采用相同的分析方法检测原药及原药加工的制剂和母药及母药加工的制剂会有一定的差异。
目前草铵膦的检测方法一般以含有少量甲醇的磷酸二氢钾水溶液或纯磷酸二氢钾水溶液作为流动相,采用液相色谱进行检测[4-6]。但实际工作中发现采用标准方法检测草铵膦原药及原药加工的制剂时效果良好,但检测母药和母药加工的制剂时,会出现无法与草铵膦有效分离的杂质峰,影响检测结果的准确性。因此,本文研究并优化了新的试验条件,用于检测草铵膦母药及母药加工而成的制剂产品中草铵膦,可以排除杂质的干扰。该方法方便、高效、准确,是一种理想的分析方法,可用于草铵膦母药、母药加工的制剂和其他草铵膦产品的检测。
1 材料与方法
1.1 供试药剂和试剂
草铵膦标准品[质量分数99.0%,国家农药质检中心(沈阳)];50%草铵膦母药;200 g/L 草铵膦水剂;磷酸二氢钾(分析纯,天津市津东天正精细化学试剂厂);磷酸(85%,成都市科隆化学品有限公司);超纯水(自制)。
1.2 主要仪器
安捷伦1260 高效液相色谱仪(配DAD 检测器,美国安捷伦科技有限公司);AB204-S 电子天平(瑞士梅特勒-托利多集团);过滤器:滤膜孔径0.45 μm。
1.3 色谱检测条件
ZORBAX SAX 不锈钢色谱柱[5.0 μm,250 mm ×4.6 mm(i.d.)];流动相:磷酸+磷酸二氢钾(质量比约1.96∶1)的水溶液;流速:1.0 mL/min;柱温:30 ℃;检测波长:195 nm;进样体积:10 μL。采用外标法,峰面积定量。在该色谱条件下,草铵膦的保留时间约为6.5 min。典型色谱图如图1 所示。

图1 相同条件下草铵膦标样、草铵膦母药和草铵膦水剂的液相色谱图
1.4 检测步骤
1.4.1 流动相配制
准确称取磷酸溶液(3.34±0.01)g,磷酸二氢钾(1.7±0.01)g,溶解于1 000 mL 超纯水中,超声脱气后备用。
1.4.2 标准溶液配制
准确称取草铵膦标准品约0.05 g(精确至0.000 2 g),置于50 mL 的容量瓶,用水溶解,超声至完全溶解后,放置至恢复室温后稀释至刻度,摇匀备用。
1.4.3 试样溶液配制
准确称取一定量(折算草铵膦含量约0.05 g)的草铵膦母药或水剂样品(精确至0.000 2 g),置于50 mL的容量瓶,用水溶解,超声至完全溶解后,放置至恢复室温后稀释至刻度,摇匀备用。
1.4.4 进样分析
按1.3 节所述的仪器条件运行液相色谱仪,观察至基线平稳、无明显波动后,对标准溶液进样分析,当连续2 次进样的草铵膦检测信号值变化小于1.5%后,视为仪器完全稳定,可进行样品的测定。按照标准溶液、试样溶液、试样溶液、标准溶液各1 针的顺序检测其中草铵膦的含量。
1.4.5 结果计算
利用工作站对草铵膦峰进行积分,并分别将标准溶液和试样溶液的峰面积平均,待测样中草铵膦质量分数w(%)计算公式如下:
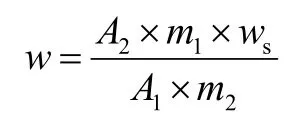
式中:A1为标准溶液铵膦峰面积平均值;A2为试样溶液草铵膦峰面积平均值;m1为称取草铵膦标准品的质量;m2为称取草铵膦待测样品的质量(g);ws为标准品中草铵膦的纯度(%)。
2 结果与讨论
2.1 检测波长的选择
采用二极管阵列检测器(DAD)在190~400 nm吸收波长范围内观察草铵膦的紫外吸收曲线,扫描结果如图2 所示。从图2 可知,在扫描范围内草铵膦的最大吸收波长为190nm,结合出峰峰形及干扰峰情况,最终选择195 nm 作为本方法的检测波长。
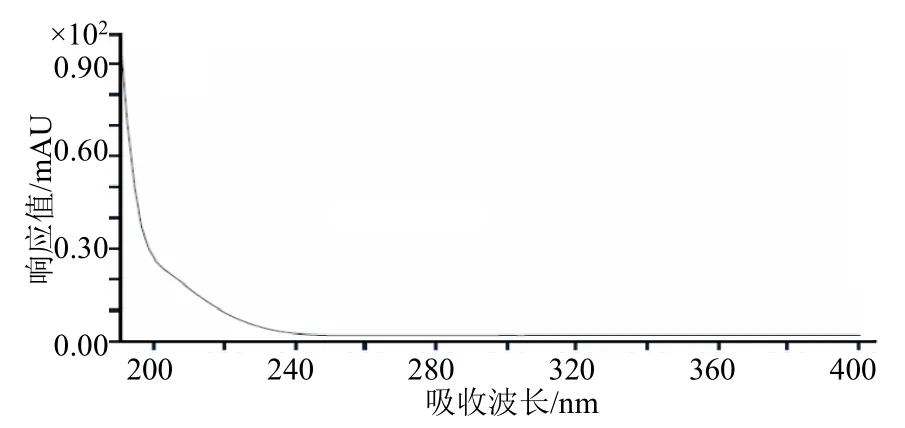
图2 草铵膦的紫外吸收光谱图
2.2 流动相的选择
目前国标和行标方法检测草铵膦采用的流动相是将磷酸二氢钾在水中溶解后,加入少量的甲醇配制而得,磷酸二氢钾的摩尔浓度为0.05 mol/L。但在实际试验操作中发现,在该流动相条件下,草铵膦母药和部分草铵膦制剂(推测为草铵膦母药而非原药加工而成)中出现了更多的杂质峰,部分杂质峰与草铵膦峰的出峰位置接近甚至重叠,无法达到良好的分离效果,从而影响检测结果的可靠性(图3)。因此本文优化了流动相条件,采用磷酸和磷酸二氢钾的缓冲体系作为流动相对草铵膦和杂质进行有效分离。试验结果发现,将含有1.7 g 磷酸二氢钾的1 000 mL超纯水(0.012 5 mol/L)中加入2.84 g 磷酸作为流动相,所得色谱图显示能够将草铵膦和杂质进行更好地分离,且峰形更好(图1),因此本试验选用该体系作为流动相。

图3 采用国标方法测定草铵膦母药和制剂的液相色谱图
2.3 分析方法的线性相关性
分别配制浓度为0.215 3、0.837 1、1.132 6、1.315 5、2.391 8 mg/mL 的草铵膦标准溶液,按照1.3 节所述色谱条件进行测定。以草铵膦的质量浓度为横坐标,峰面积为纵坐标,拟合线性回归方程,结果如图4所示。在选定质量浓度范围内,草铵膦的质量浓度与峰面积线性关系良好,相关系数(R2)为0.999 8,线性方程为y=506.57x-1.882 7。
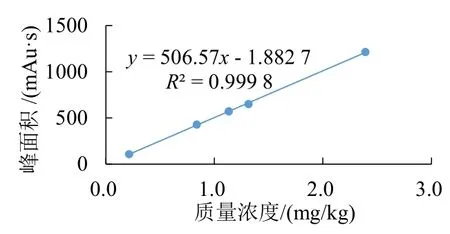
图4 线性相关试验的线性回归图
2.4 分析方法精密度
对同一草铵膦母药和水剂进行5 次平行测定,测得草铵膦的质量分数,用以计算方法的精密度,试验结果见表1。由表1 可见,草铵膦母药的标准偏差为0.081 0,变异系数为0.16%,草铵膦水剂的标准偏差为0.018 8,变异系数为0.10%,结果明显低于horwitz 公式计算的理论变异系数,说明该方法的精密度符合分析要求。

表1 精密度计算数据
2.5 方法准确度
称取已知质量分数的草铵膦母药和草铵膦水剂样品5 份,分别加入一定量的草铵膦标准品,混匀后按照前文所示方法检测溶液中草铵膦的含量,计算回收率,结果见表2。
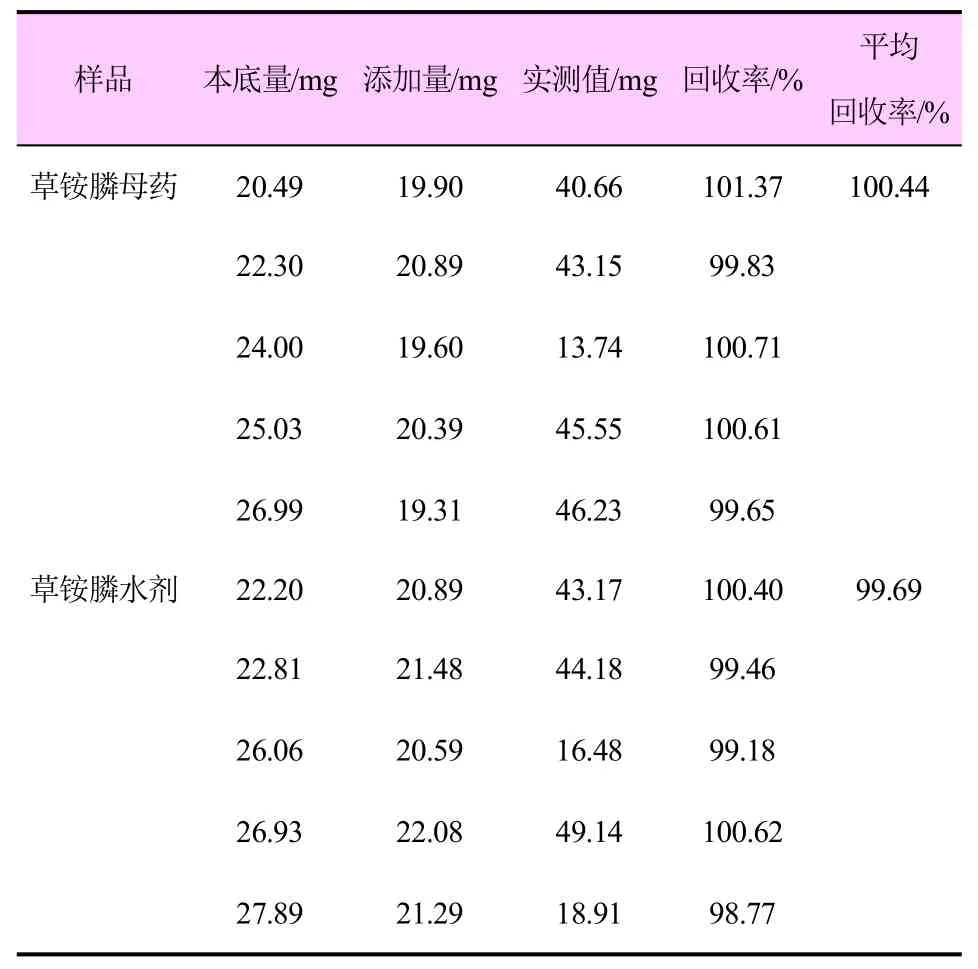
表2 分析方法的准确度试验结果
由表2 可知,该方法用于测定草铵膦母药和草铵膦水剂中草铵膦的平均回收率分别为100.44%和99.69%,符合CIPAC 的基本要求。
3 结 论
本文采用高效液相色谱仪建立了一种检测草铵膦母药和水剂有效成分的定量分析方法,其用于测定草铵膦母药和水剂中草铵膦的含量,能够有效避免杂质干扰,同时具有简便快速、重现性好、准确度高及分离效果好的优点,是草铵膦母药和母药加工的制剂中有效成分质量分数的有效检测方法,适用于此类产品的质量控制和检测。
