雷公藤内酯醇调控Nrf2/ARE通路对急性心肌梗死大鼠心肌纤维化的影响
2022-12-07熊纭辉邓海华
熊纭辉,邓海华,王 娟
(1深圳市宝安区福永人民医院心血管内科,广东 深圳 518101,2桂林医学院药学院,广西 桂林 541199)
急性心肌梗死(Acute myocardial infarction,AMI)是常见的缺血性心脏病之一,由动脉内壁上形成斑块引起的,导致流向心脏的血流量减少,并因缺氧而损伤心肌。AMI可导致心肌纤维化的发生,导致有害的心脏重塑和心脏功能障碍[1]。目前通过再灌注治疗最大限度地缩短缺血时间是降低发病率和死亡率的重要策略,再灌注可能会导致心肌缺血再灌注损伤[2]。因此,开发新的干预策略对AMI的治疗有重要意义。核因子E2相关因子2(Nuclear factor E2 related factor 2,Nrf2)是一种转录因子,可诱导大量细胞保护和抗氧化基因的表达,Nrf2与抗氧化反应元件(Antioxidant response element,ARE)结合是机体防御氧化应激的内在机制[3]。增强Nrf2信号通路可抑制心肌纤维化,减轻心肌梗死后的不良心脏重塑[4-5]。雷公藤内酯醇(Triptolide,TPL)是从中药雷公藤中提取的主要活性成分,具有抗炎、抗氧化应激、抗肿瘤等作用[6]。研究发现,TPL对缺氧复氧条件下的心肌细胞损伤有保护作用,且对肺纤维化损伤有抑制作用[7]。但TPL对AMI心肌纤维化损伤是否有抑制作用,还未见报道。本研究采用结扎冠状动脉左前降支的方法建立大鼠AMI模型,从Nrf2/ARE通路方面,探究TPL对AMI心肌纤维化损伤是否有抑制作用,现报道如下。
1 材料与方法
1.1 仪器动物ECG心电图系统(美国Nasiff Associates公司);Vevo 2100超高分辨率小动物彩色多普勒超声实时影像仪(加拿大Visual Sonics公司);iMark680多功能酶标仪(美国Bio-Rad公司);IX73倒置荧光显微镜(日本Olympus公司);H7650透射电子显微镜(日本日立公司)。
1.2 试药TPL(上海古朵生物科技公司,批号:GD-SH125-415);大鼠心肌损伤标志物乳酸脱氢酶(Lactate dehydrogenase, LDH,批号:ml003416),肌酸激酶同工酶MB(Creatine kinase isoenzyme MB,CKMB,批号:ml059533),肌钙蛋白I(Cardiac troponin I, cTnI,批号:ml059111),谷草转氨酶(Glutamic-oxalacetic transaminase,GOT,批号:ml059334),ELISA试剂盒均购自上海酶联生物科技有限公司。兔抗大鼠抗体Nrf2(批号:ab31163),转化生长因子-β1(Transforming growth factor-β1,TGF-β1,批号:ab179695),信号转导蛋白(Smad2/3,批号:ab202445),血红素氧合酶-1(Heme oxygenase 1,HO-1,批号:ab52941),超氧化物歧化酶2(Superoxide dismutase 2,SOD2,批号:ab68155),谷胱甘肽过氧化物酶(Glutathione peroxidase,GPx,批号:ab256475),α平滑肌肌动蛋白(Apha-smooth muscle actin,α-SMA,批号:ab08424),Ⅰ型胶原(Type I collagen,COL-Ⅰ,批号:ab182744),核苷酸结合寡聚化结构域样受体蛋白3(Nucleotide-binding oligomerization domain-like receptor protein 3,NLRP3,批号:ab263899),白细胞介素-6(Interleukin-6,IL-6,批号:ab208113)、白细胞介素-1β(Interleukin-1β,IL-1β,批号:ab254360)均购自英国Abcam公司。
1.3 实验动物清洁级雄性Wistar大鼠,体质量230±10 g,购自重庆大清生物有限公司,许可证号:SCXK(渝)2020-0002。本研究经福永人民医院动物伦理委员会批准同意,批号为IACUC-01(202002203)。
1.4 方法
1.4.1 大鼠急性心肌梗死(acute myocardial infarction,AMI)模型的建立 使用戊巴比妥钠(50 mg/kg)腹腔注射麻醉大鼠,仰卧位固定,连接肢体心电图电极,监测心肌缺血开始时的典型的心电图变化。沿左侧第4、5肋间切开皮肤,打开胸腔暴露心脏,使用止血钳将左心耳提起,找到冠状动脉左前降支,并结扎。心电图显示ST段弓背上抬,提示模型构建成功。手术完成后,逐层缝合大鼠肌肉和皮肤,切口处注射少量青霉素防止创面感染。
1.4.2 分组与给药 大鼠造模成功后,随机分为模型组、TPL低及高剂量组、Nrf2抑制剂组、TPL+Nrf2抑制剂组,每组12只;另取12只大鼠,只穿线不结扎冠状动脉前降支,作为假手术组。TPL低、高剂量组腹腔注射给予25、50 μg/kg的TPL;Nrf2抑制剂组腹腔注射Nrf2抑制剂-全反式维甲酸(ATRA)7 mg/kg;TPL+Nrf2抑制剂组腹腔注射TLP溶液50 μg/kg的同时注射Nrf2抑制剂ATRA;假手术组与模型组腹腔注射10 mL/kg的含1% DMSO的0.9%氯化钠溶液,各组连续给药4周,1次/d。
1.5 指标检测
1.5.1 超声检测左心室功能 采用超高分辨率小动物超声影像系统测量大鼠左心室收缩末期内径(Left ventricular end-systolic diameter, LVESD)、左心室舒张末期内径(Left ventricular end-diastolic diameter, LVEDd)及心脏短轴缩短分数(Fractional shortening, FS)变化。
1.5.2 试剂盒法测血清心肌损伤相关指标 取大鼠腹主动脉血3 mL,静置后以3 000 r/min离心15 min,分离血清,按试剂盒说明书方法检测血清心肌损伤相关标志物CK-MB、GOT、LDH、cTnI变化。
1.5.3 TTC法检测心肌梗死面积 各组随机取6只大鼠,麻醉处死,取心脏,沿冠状面将其切为5片(厚度大致相同),按TTC染液说明书方法染色,Image pro6软件分析检测心肌梗死面积。
1.5.4 透射电镜观察心肌细胞结构损伤 剩余大鼠麻醉处死后,解剖取心尖区组织,剪成1 mm3的组织块儿送于电镜室处理,将左心室组织在4℃下用4%多聚甲醛和1%戊二醛中固定过夜。用增加浓度的乙醇对切片进行洗涤和脱水,然后包埋和切成超薄切片。在H7650透射电子显微镜下观察心脏切片心肌细胞结构变化。剩余左心室组织分成两部分,一部分于-80℃保存,剩余部分石蜡包埋制成5 μm的切片。
1.5.5 Masson染色观察心肌组织纤维化变化 取心肌组织石蜡切片,按Masson染色液说明书方法染色、封片后,于显微镜下观察,Image-Pro Plus 6.0图像软件分析心肌胶原容积分数。
1.5.6 免疫组织化学法检测心肌组织Nrf2、α-SMA阳性表达 取心肌组织石蜡切片,脱蜡、水化、透化后,滴加1∶200的Nrf2、α-SMA,1∶500的生物素化二抗及链霉亲和素-过氧化物酶复合物室,显微镜下观察,Image-Pro Plus 6.0图像分析阳性(红棕色)染色区域平均光密度(MOD)值。
1.5.7 DHE荧光探针法检测心肌组织ROS水平 取冰冻心肌组织,剪成100 mm3的组织块儿,包埋后,切成10 μm的冰冻切片,DHE染液室温避光染色30 min,荧光显微镜下观察DHE染色的荧光强度,并使用Image-Pro Plus 6.0软件进行量化。
1.5.8 Western Blon法检测心肌组织Nrf2及下游炎症、氧化应激、纤维化相关蛋白表达 取冰冻心肌组织,匀浆得匀浆液,提取胞浆及胞核中蛋白,测定浓度后,取50 μg蛋白进行电泳、电转膜操作,滴加1∶500的Nrf2、TGF-β1、p-Smad2/3、Smad2/3、核苷酸结合寡聚化结构域样受体蛋白3(Nucleotide-binding oligomerization domain-like receptor protein 3, NLRP3)、IL-6、IL-1β、HO-1、SOD2、GPx、COL-Ⅰ抗体及1∶800的β-actin内参抗体,于4℃孵育过夜,次日滴加1∶1 000的羊抗兔HRP二抗溶液,室温孵育2h,增强化学发光法显色,化学发光仪观察条带并拍照,Image-J软件分析条带相对灰度值。

2 结果
2.1 TPL对大鼠心功能的影响与假手术组相比,模型组大鼠LVESD及LVEDd升高,FS降低;与模型组相比,TPL低、高剂量组大鼠LVESD及LVEDd降低,FS升高,且TPL剂量越高改善越明显;Nrf2抑制剂组大鼠LVESD及LVEDd升高,FS降低;与TPL低、高剂量组相比,TPL+Nrf2抑制剂组大鼠LVESD及LVEDd升高,FS降低,差异均有统计学意义(P<0.05),见表1。

表1 大鼠心功能相关指标比较
2.2 TPL对大鼠心肌损伤标志物的影响与假手术组相比,模型组大鼠血清CK-MB、GOT、LDH、cTnI水平升高;与模型组相比,TPL低、高剂量组大鼠血清CK-MB、GOT、LDH、cTnI水平降低,且TPL剂量越高心肌损伤标志物降低越明显;Nrf2 抑制剂组大鼠血清CK-MB、GOT、LDH、cTnI 水平升高;与TPL低、高剂量组相比,TPL+Nrf2 抑制剂组上述指标变化升高,差异均有统计学意义(P<0.05),见表2。

表2 大鼠心肌损伤标志物比较
2.3 TPL对大鼠心肌梗死面积的影响与假手术组相比,模型组大鼠心肌梗死面积升高;与模型组相比,TPL低、高剂量组大鼠心肌梗死面积降低,且TPL高剂量组对心肌梗死面积降低作用优于低剂量组;Nrf2抑制剂组大鼠心肌梗死面积进一步升高;与TPL低、高剂量组相比,TPL+Nrf2抑制剂组心肌梗死面积升高,差异均有统计学意义(P<0.05),见表3。

表3 大鼠心肌梗死面积比较
2.4 TPL对大鼠心肌组织细胞超微结构的影响模型组大鼠可见细胞体积变小、胞质浓缩、胞核裂解为碎块、核内空泡结构增多,线粒体肿胀,心肌肌节断裂且排列紊乱。TPL低、高剂量组大鼠上述细胞结构损伤逐渐减轻,几乎未见空泡化的细胞核。Nrf2抑制剂组大鼠细胞核碎裂、空泡化及心肌肌节断裂等结构损伤进一步加重。TPL+Nrf2抑制剂组心肌结构损伤变化与模型组相近,见图1。

假手术组 模型组 TPL低剂量组
2.5 TPL对大鼠心肌纤维化的影响模型组大鼠心肌排列紊乱,胶原沉积增加,心肌纤维消失,局灶性梗死化和瘢痕形成明显,心肌组织胶原纤维蓝染加重,α-SMA呈强阳性(红棕色)表达,胶原容积分数、α-SMA阳性表达均高于假手术组。与模型组相比,TPL低、高剂量组大鼠胶原容积分数、α-SMA阳性表达降低,且TPL剂量越高心肌心肌纤维化改善越明显;Nrf2抑制剂组大鼠胶原容积分数、α-SMA阳性表达较模型组进一步升高;与TPL低、高剂量组相比,TPL+Nrf2抑制剂组大鼠胶原容积分数、α-SMA阳性表达升高,差异均有统计学意义(P<0.05),见图2、表4。

Masson染色α-SMA假手术组模型组TPL低剂量组TPL高剂量组Nrf2抑制剂组TPL+Nrf2抑制剂组
表4 大鼠心肌组织胶原容积分数、α-SMA阳性表达比较
2.6 TPL对大鼠氧化应激的影响假手术组大鼠心肌组织中ROS荧光强度较弱,几乎未见呈红色荧光的ROS;模型组大鼠心肌组织ROS荧光强度升高,抗氧化应激蛋白HO-1、SOD2、GPx蛋白表达升高;与模型组相比,TPL低、高剂量组大鼠ROS荧光强度降低,抗氧化应激蛋白HO-1、SOD2、GPx蛋白表达进一步升高,且TPL剂量越高上述指标改善越明显;Nrf2抑制剂组大鼠ROS荧光强度升高,抗氧化应激蛋白HO-1、SOD2、GPx蛋白表达降低;与TPL各剂量组相比,TPL+Nrf2抑制剂组ROS荧光强度升高,HO-1、SOD2、GPx蛋白表达降低,差异均有统计学意义(P<0.05),见图3、表5。
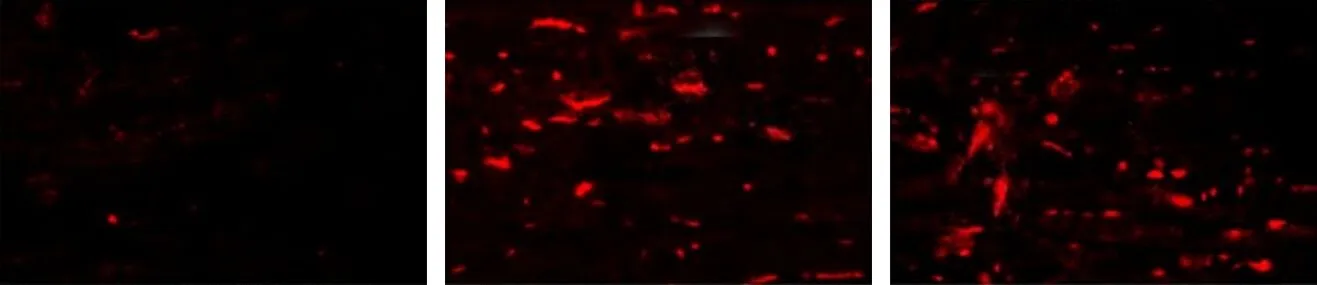
假手术组 模型组 TPL低剂量组
表5 大鼠ROS荧光强度及蛋白表达比较〗n=6)
2.7 TPL对大鼠炎症反应的影响模型组大鼠心肌组织NLRP3、IL-6、IL-1β蛋白表达显著高于假手术组;与模型组相比,TPL低、高剂量组大鼠心肌组织NLRP3、IL-6、IL-1β蛋白表达降低,且TPL剂量越高上述指标改善越明显;Nrf2抑制剂组大鼠心肌组织NLRP3、IL-6、IL-1β表达较模型组进一步升高;与TPL低、高剂量组相比,TPL+Nrf2抑制剂组NLRP3、IL-6、IL-1β表达升高(P<0.05),差异均有统计学意义,见表6。
表6 大鼠心肌组织NLRP3、IL-6、IL-1β蛋白表达比较
2.8 VTPL对大鼠Nrf2及下游TGF-β1、1-Smad2/3、COL-Ⅰ等促纤维化相关蛋白表达的影响模型组大鼠Nrf2阳性(红棕色)表达于心肌细胞胞核中,Nrf2及下游TGF-β1、(p-Smad2/3)/(Smad2/3)、COL-Ⅰ表达显著高于假手术组;与模型组相比,TPL低、高剂量组大鼠Nrf2阳性表达及蛋白表达升高,TGF-β1、(p-Smad2/3)/(Smad2/3)、COL-Ⅰ促纤维化蛋白表达降低,且TPL剂量越高上述指标改善越明显;Nrf2抑制剂组大鼠Nrf2阳性表达及蛋白表达降低,TGF-β1、(p-Smad2/3)/(Smad2/3)、COL-Ⅰ促纤维化蛋白表达升高;与TPL低、高剂量组相比,TPL+Nrf2抑制剂组大鼠Nrf2阳性表达及蛋白表达降低,TGF-β1、(p-Smad2/3)/(Smad2/3)、COL-Ⅰ促纤维化蛋白表达升高,差异均有统计学意义(P<0.05),见图4,表7。
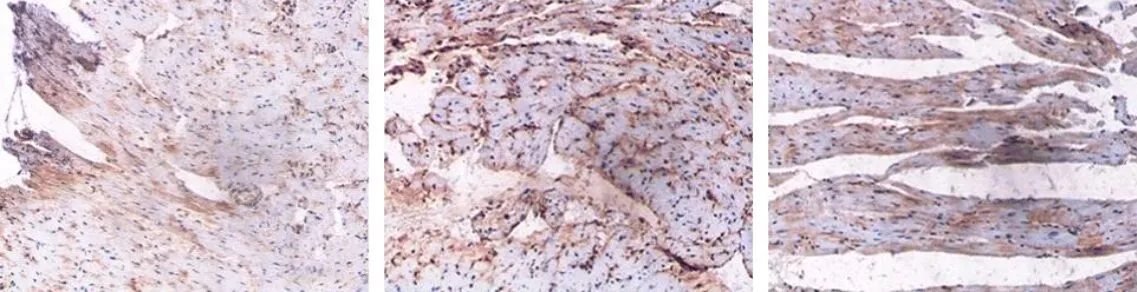
假手术组 模型组 TPL低剂量组
表7 大鼠心肌组织Nrf2及下游TGF-β1、Smad2/3、COL-Ⅰ蛋白表达比较
3 讨论
AMI已被证明会导致慢性进行性心脏的不良重塑,主要涉及心脏纤维化、炎症、氧化应激和心肌细胞凋亡,并最终导致心脏功能障碍[8-9]。cTnI通常不存在于血液中。当心肌细胞受损时,肌钙蛋白从心肌纤维中释放出来,并从心肌细胞扩散到血液循环中。cTnI的升高是对心肌坏死最特异性和最敏感的反应。此外,临床上用于心肌梗死诊断的常用酶指标包括GOT、CK-MB和LDH,这些指标的升高也可以不同程度地反映心肌损伤[10]。在本研究中,笔者观察到模型大鼠血清中心肌损伤标志物cTnI、GOT、CK-MB和LDH增加,且LVESD、LVEDd升高,FS降低,心肌梗死面积增加,这与既往的研究结果一致[11]。提示模型大鼠出现心肌损伤和心脏功能障碍。
据报道,AMI心肌细胞受到缺血缺氧刺激,可产生大量ROS而破坏细胞基质,引起心肌组织氧化应激损伤[12];并且ROS也是促进心肌成纤维细胞过度增殖及分泌胶原蛋白的重要因子[13]。此外,炎症也参与AMI后心肌纤维化的发生、发展。在心肌梗死的急性期,炎性细胞因子TNF-α、IL-1β、IL-6和转化生长因子(如TGF-β1)的表达升高,随后会诱导胶原沉积[14]。本研究在模型大鼠梗死组织中检测到大量ROS生成,抗氧化酶表达降低以及NLRP3、IL-6、IL-1β、TGF-β1、胶原蛋白COL-Ⅰ、α-SMA表达的升高,提示氧化应激和炎症反应参与AMI后心肌纤维化的发生。由于氧化应激和炎症在AMI后的心脏重塑中发挥重要的病理生理作用,因此抗氧化剂和抗炎疗法可能对心肌损伤有益。
TPL是一种源自雷公藤的活性化合物,已被证实具有心脏保护作用,且其保护机制与抗氧化应激和抗炎作用有关。研究显示,在大鼠心肌缺血再灌注模型中,TPL可通过激活Nrf2/HO-1信号通路,抑制再灌注心肌组织中TNF-α、IL-1β、IL-6和丙二醛的过量产生,并上调抗氧化SOD、GSH和GPx活性,发挥心脏保护作用[15]。此外,NLRP3炎症小体被认为是调节炎症的关键因子[16]。在异丙肾上腺素诱发的心肌纤维化大鼠模型中,TPL可以通过抑制NLRP3炎症小体的激活,下调IL-1β和TNF-α的表达来改善心肌纤维化并下调纤维化相关因子(TGF-β1、COL-Ⅰ)的表达[17]。Mehdipoor等[18]发现炎症及氧化应激可促进TGF-β1-Smad2/3通路活化,促进心肌上皮细胞向间质转化而加剧心肌纤维化发展。在本研究中,TPL干预有效降低了血清cTnI、GOT、CK-MB和LDH水平以及炎症因子NLRP3、IL-6、IL-1β的表达,增加了抗氧化应激蛋白HO-1、SOD2、GPx表达,且降低了ROS的产生和TGF-β1、p-Smad2/3、COL-Ⅰ表达;表明TPL可抑制炎症和氧化应激,并抑制TGF-β1-Smad2/3通路激活,减轻AMI大鼠心肌组织纤维化;这可以通过Masson染色和免疫组织化学染色中心肌组织纤维化程度降低和α-SMA阳性表达的降低来证明。
Nrf2是防御氧化应激的关键调节剂之一。Nrf2可在形成异源二聚体后,与ARE结合并调控下游众多抗氧化蛋白表达,来抑制内源性和外源性ROS蓄积引起的细胞氧化应激损伤,抑制心肌纤维化[19-20]。另外,Nrf2还可通过与Trx1/TXNIP复合物相互作用,来激活细胞内保护程序、抑制炎症小体NLRP3的活化、降低细胞内IL-6、IL-β1等表达,发挥抗炎反应[21]。Xu等[22]人的研究显示,激活Nrf2/ARE通路可减少炎性细胞因子的释放和NLRP3的激活,增加抗氧化酶的活性,抑制氧化应激和炎症反应来改善链脲佐菌素诱导的糖尿病小鼠心肌纤维化。本研究发现,模型组大鼠心肌组织中Nrf2阳性表达升高,用Nrf2/ARE通路抑制剂ATRA阻断Nrf2通路活化后,大鼠心肌组织氧化应激、炎症反应进一步加重,大鼠心肌梗死面积及纤维化损伤最严重,提示Nrf2/ARE介导的抗炎、抗氧化反应,可能是机体发挥抗AMI纤维化损伤的关键机制。TPL被报道可以通过激活Nrf2通路来抑制炎症和氧化应激,减少深低温停循环引起的脑损伤[23]。在本研究中,经TPL处理的大鼠心肌组织中Nrf2介导的抗炎、抗氧化反应进一步增强,心肌纤维化减轻,提示TPL可能通过促进Nrf2/ARE介导的抗炎、抗氧化反应活化,来缓解AMI大鼠心肌坏死及纤维化发展。为了验证TPL的心脏保护作用是否与Nrf2/ARE通路有关,本研究在TPL处理的基础上,给予Nrf2/ARE通路抑制剂ATRA进行干预,发现ATRA可明显逆转TPL对AMI大鼠心肌纤维化的抑制作用。提示TPL可能通过激活Nrf2/ARE通路,抑制氧化应激和炎症反应,减轻AMI大鼠心肌纤维化。
综上所述,TPL可激活Nrf2/ARE抗氧化、抗炎反应途径,改善AMI大鼠心肌坏死及纤维化发展,最终改善心脏功能。本研究为TPL治疗AMI提供一定的理论依据。但自噬、能量代谢、免疫等功能异常,也与心肌纤维化发展关系密切,TPL能否通过上述途径改善AMI纤维化发展,还有待后续继续研究。
