A natural manganese ore as a heterogeneous catalyst to effectively activate peroxymonosulfate to oxidize organic pollutants
2022-12-07JinhunGuPingYinYiChenHonglinZhuRuiWng
Jinhun Gu, Ping Yin, Yi Chen,∗, Honglin Zhu, Rui Wng
a School of Food and Bioengineering, Civil Engineering and Architecture and Environment, Emergency Science, Xihua University, Chengdu 610039, China
b Sichuan Rongxinkai Engineering Design Co., Ltd., Chengdu 610039, China
c Faculty of Geosciences and Environmental Engineering, Southwest Jiaotong University, Chengdu 611756, China
Keywords:Transition metal Heterogeneous catalyst Natural ore Orange II Peroxymonosulfate
ABSTRACT Heterogeneous transition metal catalysts are indispensable in improving environmental pollution.However, their fabrication is often costly and cumbersome, and they can easily pollute the environment.This study proposed using a natural Gabonese ore (GBO) containing MnxOy and FexOy as catalysts to degrade orange II (OII) via peroxymonosulfate (PMS) activation.The GBO + PMS system exhibited extraordinarily high stability and catalytic activity towards OII elimination (92.2%, 0.0453 min−1).The reactive oxygen species (ROS) generated in the system were identified using radical scavenging tests and electron spinresonance (ESR) analysis.Singlet oxygen (1O2) represented the dominant reactive species for OII degradation, while the system presented a lower reaction energy barrier and was effective in a broad pH range(2–10).This work also proposed the activation mechanism for the GBO + PMS system and OII degradation pathways.This study revealed a new approach for exploring inexpensive, eco-friendly, efficient, and stable heterogeneous transition metal catalysts.
Advanced oxidation processes (AOPs) are effective in completely decomposing target organic pollutants in wastewater [1–5].Sulfate radical (SO4•−)-based AOPs (SR-AOPs) show significant promise for treating the refractory organics in water [6–8] since SO4•−exhibits a higher standard redox potential (E0= 2.5–3.1 Vvs.1.8–2.7 V of•OH) and a longer half-life period (30–40 μsvs.20 ns of•OH) in a wide pH range [6,9–13].SO4•−mainly results from persulfate (PS)or peroxymonosulfate (PMS) activation.In addition, it is believed that PMS is more easily dissociated into SO4•−than PS due to its asymmetric structure and lower bond energy of “O−O” [7,14,15].Various methods have been developed for PMS stimulation to generate SO4•−, such as heat [16,17], ultrasound [18,19], and ultraviolet(UV) light [20,21].However, using these methods to activate PMS requires additional energy, which is expensive.Therefore, identifying a simple and economical method for PMS activation is essential.
Heterogeneous catalytic activation is considered promising since the heterogeneous catalyst is easily separated and reused without requiring additional energy [10,22].Furthermore, transition metals, oxides, and bimetallic oxides are reportedly efficient heterogeneous catalysts for PMS activation since they necessitate less complex system configurations and are more economical for organic pollutant decontamination [6,10].Although recent studies have shown that cobalt represents the most effective PMS activator [7,23,24], it is expensive and leads to unavoidable, highly toxic leaching.Therefore, it is essential to explore non-cobalt-based heterogeneous catalysts for PMS application during environmental remediation.
The performance of heterogeneous transition metal-based catalysts heavily depends on the nature of the transition metal, the valence state, crystallinity, structure, and surface morphology [1,25].Due to the natural abundance of multivalent transition metals,iron oxides, and manganese oxides, they are more environmentally friendly than cobalt ions and possibly provide an alternative to cobalt-based catalysts.Mixed metal catalyst loading reportedly improves redox activity and catalytic capacity due to the synergistic redox coupling of different metals and the presence of mixedvalence transition metals [6,10,26].Consequently, iron-manganese bimetallic oxides have been synthesized to further enhance their catalytic capacity and have exhibited a significantly higher catalytic capacity than monometallic oxides [26–29].Although considerable advances have been made, challenges remain regarding the fabrication of iron-manganese bimetallic oxide catalysts.The process is complicated, costly, and can easily cause environmental pollution.Therefore, developing a simple, inexpensive, eco-friendly technique for catalyst synthesis or exploring natural materials exhibiting catalytic bimetallic oxide properties is crucial.
This study proposes a natural manganese ore containing iron and manganese elements as a PMS activation catalyst while evaluating its catalytic capacity and stability.The catalyst is characterized systematically and is employed for PMS decomposition with orange II (OII) as the representative pollutant.A series of catalytic tests are performed to identify the main conditions affecting the catalytic capacity and the main reactive species.Based on the results, a simple mechanism for PMS decomposition and the subsequent degradation and mineralization of OII is proposed.
The X-ray diffraction (XRD) pattern (Fig.1a) showed thatδ-MnO2(JCPDS No.80-1098), Mn3O4(JCPDS No.13-0162), Fe3O4(JCPDS No.89-0688), and SiO2(JCPDS No.46-1045) represented the primary GBO phases, while the manganese oxide content was the highest (Fig.S1 in Supporting information).The scanning electron microscopy (SEM) images in Fig.1b showed that GBO consisted of different-sized particles, appearing as piles of layered structures mainly related toδ-MnO2[30,31].The N2adsorptiondesorption isotherms of GBO and GBO–OII were type V with H3 hysteresis loops, as depicted in Fig.S2 (Supporting information).The BET surface area (SBET) of GBO was 27.39 m2/g, and the total pore volume (Vtot) was 0.054 cm3/g (Table S1 in Supporting information), mainly denoting mesoporous characteristics.The XRD pattern, SEM, and textural properties of GBO–OII showed minor changes after catalyzation, indicating that the GBO structure was relatively stable.
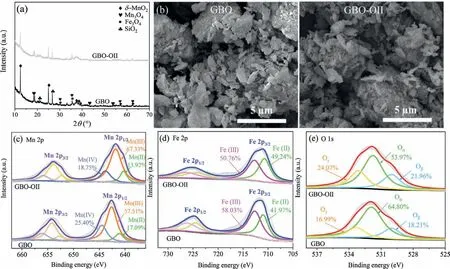
Fig.1 .(a) The XRD patterns and (b) SEM images of the GBO and GBO–OII, and XPS spectra of the GBO catalyst before and after the reaction: (c) Mn 2p, (d) Fe 2p, and (e)O 1s.
Wide scan X-ray photoelectron spectroscopy (XPS) spectra (Fig.S3a in Supporting information) were used to investigate the elemental composition and chemical status of Mn, Fe, and O in the GBO catalyst.This indicated that GBO primarily consisted of O, Mn,Fe, Si, and Al, corresponding to its main elemental composition.The Mn 2p3/2spectra in the fresh GBO (Fig.1c) presented three individual peaks at 641.1 eV, 642.6 eV, and 644.5 eV, which were assigned to Mn(II), Mn(III), and Mn(IV), respectively [1], indicating the polyvalent state of Mn in GBO.The Fe 2p3/2peaks (Fig.1d)at 710.9 eV and 712.6 eV corresponded to Fe(II) and Fe(III), respectively [32–35].As illustrated in Fig.1e, the O 1s spectra of the fresh and used GBO were deconvoluted into three components: lattice oxygen (Oβ, O2−), surface oxygen (Oα, −OH), and adsorbed oxygen (Oγ, H2O) at 530.0 eV, 531.9 eV, and 533.3 eV, respectively[7,36,37].The −OH groups can reportedly provide active sites for PMS bonding [2,37,38].A reduction of 10.83% was evident in the oxygen content on the GBO surface after the reaction, indicating that −OH was involved in the reaction process, which was also confirmedviaFourier transform infrared spectroscopy (FTIR) (Fig.S3b in Supporting information).Furthermore, −CH2signal peaks at 2989 cm−1and 2901 cm−1were observed on GBO–OII [39], indicating OII decomposition on the GBO surface.In general, the FTIR spectra of GBO changed slightly, indicating the relative stability of the GBO surface properties.
A series of experiments were performed to investigate the catalytic oxidation ability.In all cases, the degradation process of OII followedpseudo-first-order kinetics, while thepseudo-first-order rate constant (kobs) was acquiredviathe linear regression of ln(Ct/C0) to time (Eq.1) [1,40,41].

wheretis the reaction time, andC0andCtare the OII concentrations at initial andttime.
The OII removal was evaluated in various aqueous systems, and the results are shown in Fig.2a.The adsorption of OII on GBO and PMS alone was negligible.It is well known that PMS hydrolysis generates abundant H+, further influencing the initial pH of the solution [41].Considering that the pH of the aqueous system changed(about 3.84) after adding PMS, the catalytic oxidation properties of GBO were investigated at a solution pH of 3.84.The OII removal rate in the GBO (pH 3.84) (11.7%) system was noticeably higher than GBO (6.5%) and PMS (2.7%).
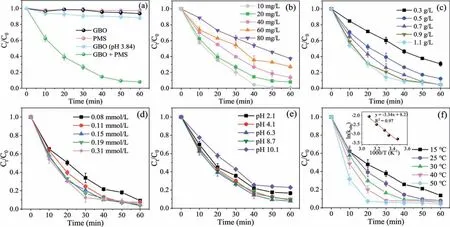
Fig.2 .(a) The degradation of OII in various systems (MGBO = 0.9 g/L).The impact of (b) the initial OII solution concentration (MPMS = 0.14 mmol/L), (c) GBO dosage,(d) PMS dosage (MGBO = 0.9 g/L), (e) initial pH (MPMS = 0.14 mmol/L), and (f) reaction temperature on OII degradation.Conditions: C0 = 20 mg/L, MGBO = 0.97 g/L,MPMS = 0.15 mmol/L, pH 6.6, T = 25 °C.
GBO (pH 3.84) alone only induced an OII removal of 11.7% in 60 min, while a 92.2% rate was achieved by the GBO + PMS system, exceeding the total of that in the PMS (2.7%) and GBO(pH 3.84) (11.7%) systems, indicating a synergistic effect between PMS and GBO.In addition, the reaction rate constantkobsvalues were further compared (Fig.S4a in Supporting information).Thekobsvalue of the GBO + PMS system (0.0453 min−1) was 18.88 times and 41.18 times higher than in the GBO (pH 3.84)(0.0024 min−1) and GBO system (0.0011 min−1), respectively.In addition, the catalytic activity of the pure substances in GBO (δ-MnO2, Mn3O4, Fe3O4,and SiO2) was compared in Fig.S5a (Supporting information).The results show that Mn3O4displayed the strongest catalytic activity, whileδ-MnO2and Fe3O4were weak,and SiO2exhibited no activity.However, the removal rate of OII by GBO (92.2%) was substantially higher than Mn3O4(27.0%).These results suggest that GBO displays a significant capacity for PMS activation and OII elimination.Furthermore, GBO demonstrates excellent removal performance involving other dyes in wastewater (Fig.S5b in Supporting information), highlighting its promise for practical application.
The parameters affecting OII oxidation were investigated, including the OII concentration (C0), GBO dosage (MGBO), PMS dosage(MPMS), initial solution pH, and reaction temperature (T).The OII was completely removed within 60 min at an initial concentration of 10 mg/L (Fig.2b).As theC0increased, the OII removal rate and the reaction rate constant (kobs, Fig.S4b in Supporting information)decreased significantly.In addition, the removal rate andkobsincreased in conjunction withMGBO(Fig.2c).However, at dosages of 0.9 g/L and 1.1 g/L, the respective systemkobswere close (Fig.S4c in Supporting information), indicating complete PMS decomposition by the catalyst.It is well known that the PMS concentration significantly affects catalytic reactions.Fig.2d and Fig.S4d (Supporting information) verify that a high PMS dosage may generally increase the reaction rate.However, it has been proposed that PMS overdosage has a scavenging effect on SO4•−(Eq.2), while excessive SO4•−can also cause a self-scavenging reaction (Eq.3) [7,42].Therefore, 0.15 mmol/LMPMSis more appropriate for OII degradation.
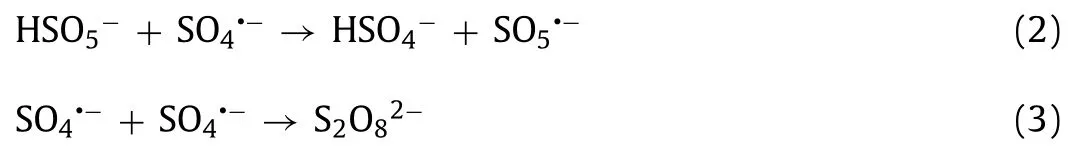
The solution pH plays a crucial role in the heterogeneous catalytic oxidation process by influencing substrate and oxidant speciation [41,43].As illustrated in Fig.2e, the removal of OII was inhibited slightly in acidic and alkaline conditions, with only about 83.5% and 77.16% of OII removed at pH 2.1 and pH 10.1, respectively.In acidic conditions, the high H+concentration scavenged•OH and SO4•−, reducing OII degradation (Eqs.4 and 5) [44–46].In alkaline conditions, the PMS became unstable, decomposing to sulfate ions (SO42−) and oxygen (O2) (Eq.6) [44], consequently reducing the OII degradation rate.The GBO + PMS system achieved optimal OII removal at a pH of 6.3, with the removal rate reaching 92.2% at akobsof 0.0472 min−1.Moreover, the OII removal rate(>91.2%) andkobs(>0.0413 min−1) were relatively high at pH values ranging from 4.1 to 8.7.The broad pH application range indicates that the established GBO + PMS system exhibits significant potential for OII degradation.
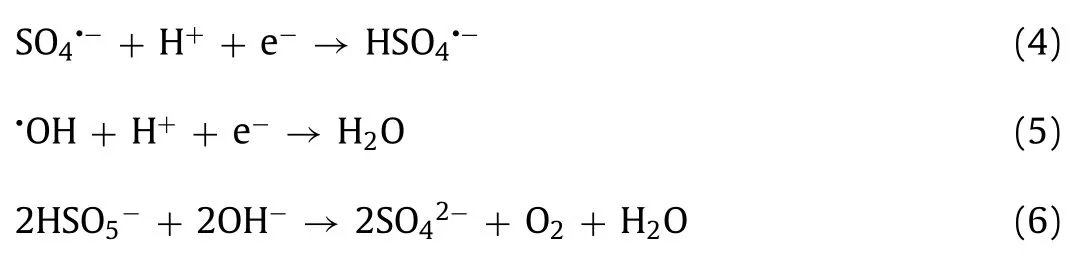
As shown in Fig.2f and Fig.S4f (Supporting information), the OII removal and reaction rates increased significantly in conjunction with higher temperatures.The OII removal rate increased from 86.6% to 97.9% as the temperature rose from 15 °C to 50 °C, while thekobsvalue was rapidly elevated from 0.0372 min−1to 0.1283 min−1, indicating that the GBO activation of PMS was an endothermic reaction.The reaction produced more active sulfate and hydroxyl radicals at high temperatures, improving the OII degradation efficiency [7,47].
The activation energy (Ea) of the OII degradation in the GBO + PMS system was calculated using the Arrhenius equation(Eq.7), producing a value of 27.77 kJ/mol, significantly exceeding theEavalues in the diffusion-controlled reactions (10–13 kJ/mol)[23,32].The apparent reaction rate of OII degradation was mainly determined by the intrinsic chemical reaction rate on the GBO surface rather than the mass transfer rate [41,48].In addition, theEavalue (27.77 kJ/mol) was substantially lower than that previously reported for PMS activationviadifferent heterogeneous catalysts(Table S2 in Supporting information).This suggests that GBO with a lower reaction energy barrier can remove pollutants, rendering it promising for practical application.

whereAis the prefactor, andRis the universal gas constant(8.314 J mol−1K−1).
The effect of the inorganic anions in water on OII removal was also investigated.As shown in Figs.S5c and d (Supporting information), the introduction of Cl−(0–20 mmol/L) promoted the OII degradation process, while HCO3−(0–20 mmol/L) inhibited it.Previous studies have shown that Cl−can be oxidized by PMS to form HOCl as an additional oxidant for OII degradation [49,50].HCO3−can react with PMS to form fewer active free radicals, leading to the inefficacy of PMS [39].Therefore, although HCO3−influenced the OII degradation rate, the overall degradation efficiency remained>86.0%, indicating that the GBO + PMS system displayed excellent resistance to inorganic ions.
PMS activation can reportedly produce different types of ROS, such as SO4•−,•OH, O2•−, and1O2[1,32,41].ROS represents the main factors of OII degradation catalyzed by GBO + PMS.A series of quenching tests were performed to further investigate the key active species in the system.Ethyl alcohol (EtOH) was used as a scavenger for both SO4•−(k2(SO4•−, EtOH) = 1.6–7.7 × 107L mol−1s−1) and•OH (k2(•OH,EtOH) = 1.2–2.8 × 109L mol−1s−1), whiletert–butyl alcohol(TBA) was used as an effective quenching agent for•OH (k2(•OH,TBA) = 3.8–7.6 × 108L mol−1s−1) [7,41,42].Fig.3a illustrates that 300 mmol/L EtOH or TBA had a minimal impact on OII oxidation,while 1 mol/L EtOH or TBA enhanced its inhibition.However, about 81.8% (EtOH) and 74.1% (TBA) of OII could be eliminated, implying that only a small amount of SO4•−and•OH were produced.TBA at 1 mol/L displayed a significantly stronger inhibitive effect on OII degradation than EtOH.This could be attributed to the fact that the higher viscosity of TBA concealed the active sites on the GBO surface, reducing catalytic efficiency [32].
Furfuryl alcohol (FFA) and L-Histamine (L-His) were used to explore the presence of1O2(k2(1O2, FFA) = 1.2 × 108L mol−1s−1,k2(1O2, L-His) = 3.2 × 107L mol−1s−1), while benzoquinone (BQ)was selected for O2•−(k2(O2•−, BQ) = 0.9–1 × 109L mol−1s−1)scavenging [41,51].According to the results shown in Fig.3b, BQ had little effect on OII degradation, possibly indicating that O2•−contributed minimally to the system.Previous studies have shown that BQ can activate PMS and accelerate its decomposition to form1O2(Eqs.8 and 9) [52,53].Therefore, the inapparent inhibition of OII degradation by BQ may be due to1O2as the main reactive species promoting the catalytic process.Furthermore, 30 mmol/L L-His and FFA exhibited a significant inhibitory impact on OII oxidation.L-His completely inhibited OII oxidation (Fig.S6 in Supporting information,k30mmol/L,L-His= 0.0006 min−1), demonstrating that1O2was generated in the system, denoting the dominant reactive species for the degradation of OII.This phenomenon indicated that1O2represented the dominant reactive species for OII degradation, followed by•OH, SO4•−, and O2•−.
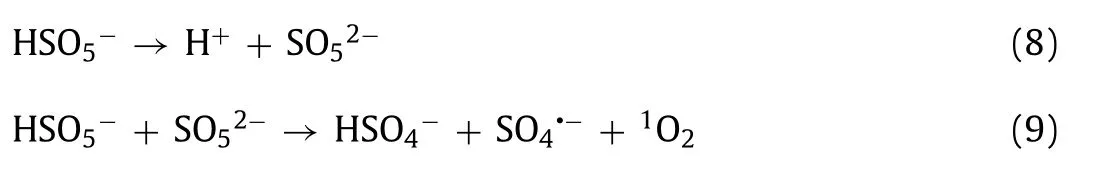
Electron spin-resonance (ESR) spectroscopy confirmed the presence of the active species in the GBO + PMS system using 5,5-dimethyl-1-pyrrolineN-oxide (DMPO) and 2,2,6,6-tetramethyl-4-piperidone (TEMP) as spin-trapping agents, as shown in Figs.3ce.Fig.3c illustrates the typical TEMP-1O2spin adduct signal observed in the PMS-only and GBO + PMS systems, which was significantly enhanced in the GBO + PMS system.DMPO-•OH/SO4•−and DMPO–O2•−adduct signals were also observed after introducing DMPO to the PMS-only and GBO + PMS systems (Figs.3d and e).Furthermore, all the samples showed similar species signals, while those at 10 min were stronger than at 5 min.The corresponding signals of the GBO + PMS system were distinctly stronger than in the PMS-only system, which might explain the enhanced PMS activation by the GBO catalyst.
GBO displayed excellent reusability for PMS activation and OII degradation in circumneutral pH conditions (initial pH 6.6).The OII degradation rate declined by only 3% after five consecutive reaction cycles (Fig.3f), which was likely due to slight Mn leaching(<0.75 mg/L, Fe leaching cannot be detected) (Fig.S6c), as well as Mn2+/Mn3+/Fe2+oxidation.
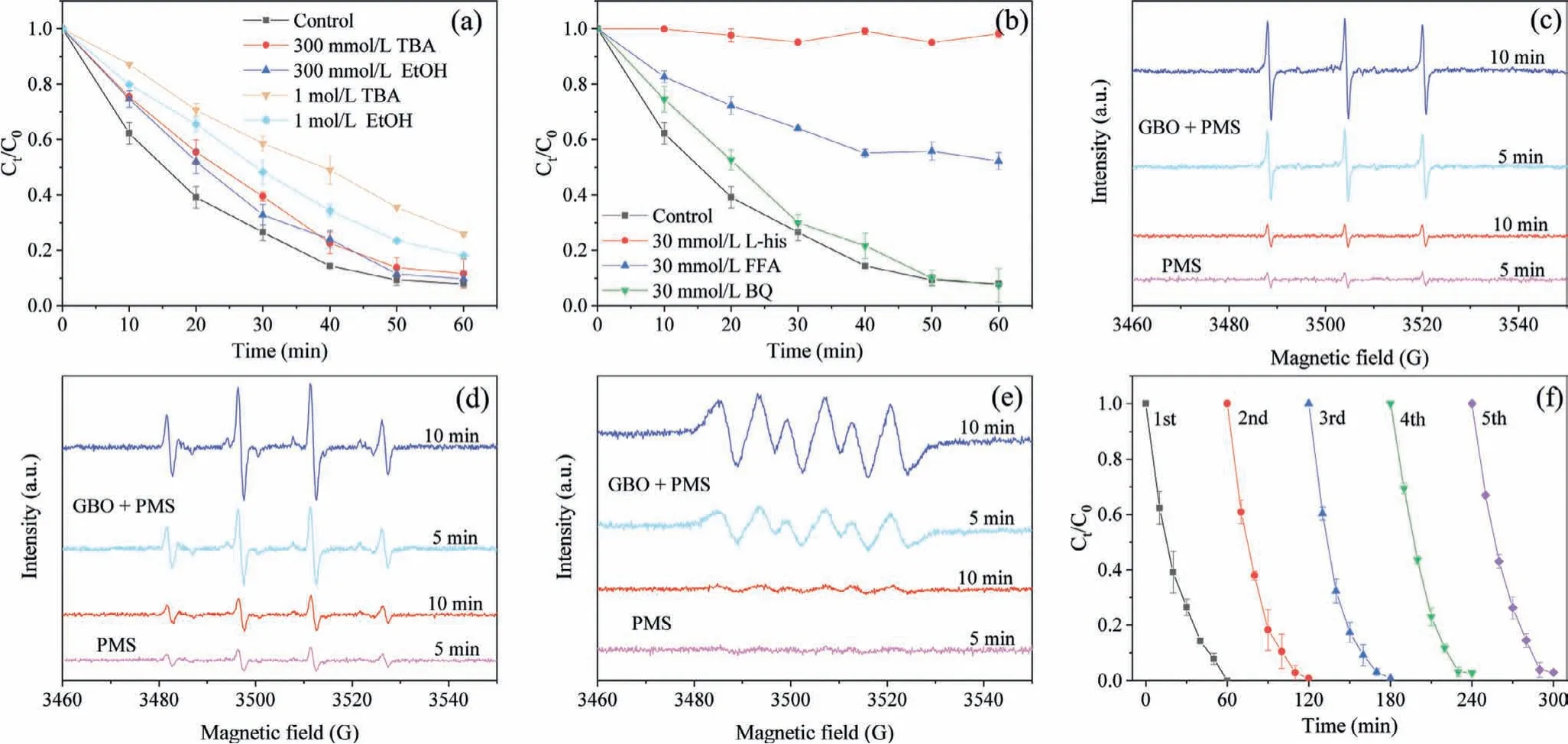
Fig.3 .(a) The quenching effect of •OH and SO4•−on OII degradation.(b) The quenching effect of 1O2 and O2•−on OII degradation.The ESR spectra of the (c) TEMP-1O2, (d)DMPO-•OH/SO4•−and (e) DMPO–O2•−adducts formed in PMS-only and GBO + PMS systems after 5 min and 10 min.(f) A recycling study of OII degradation.Conditions:C0 = 20 mg/L, MGBO = 0.97 g/L, MPMS = 0.14 mmol/L, pH 6.6, T = 25 °C.
The redox behavior of GBO in a mixed solution containing Na2SO4and PMS was studied using cyclic voltammetry (CV).As shown in Fig.S7 (Supporting information), the anode and cathode peaks indicated that redox reactions had occurred on the surface of the catalyst [2,37,54].The GBO corrosion potential was measured using Tafel polarization curves to further investigate the electron migration rate of GBO.Fig.S7b shows that the GBO exhibited a much higher corrosion current (3.83 × 10−3A) than the previously reported catalyst.This implies that the GBO demonstrates a high electron transfer rate for superior catalytic capability [7,41].
As shown in Figs.1c-e, the GBO surface redox reaction was verifiedviaXPS.The peak intensity of the GBO–OII Mn(IV) species was noticeably lower than GBO (11.48%).Mn(III) exhibited a 9.82%increase, while Mn(II) was about 1.66% higher, confirming the conversion between the two Mn species on the GBO surface.Compared with the Fe species content at various valence levels before and after the reaction, Fe(III) exhibited a 16.06% decrease since it was partially reduced to Fe(II).The results confirmed the presence of the Fe(III) and Fe(II) cycles during the reaction.Moreover, the relative content of Oαdecreased from 64.80% to 53.97%, Oβincreased from 16.99% to 24.07%, and Oγincreased by about 3.75%.These findings can be attributed to the formation of higher lattice oxygen levels on the GBO surface, while the surface −OH and H2O participated in the redox reaction of the Mn and Fe species.
Based on active radical identification and XPS analysis, a plausible GBO + PMS activation mechanism for OII degradation was proposed.HSO5−was attached to the GBO surface in an OII solution.Manganese and iron oxides with different metal center valences(Mn(II), Mn(III), Mn(IV), Fe(II), and Fe(III)) on the GBO surface acted as active HSO5−reaction sites to generate SO4•−(Eqs.10–16) [1,26,46] and•OH (Eqs.16 and 17) [7,47].Moreover, Mn(IV) reacted with Fe(II) to produce Mn(III) and Fe(III) (Eq.18) [26].The SO4•−further reacted with H2O (or OH−) to produce•OH, based on Eqs.19 and 20 [1,55].Furthermore, the HSO5−decomposed to form SO4•−,1O2, and SO52−(Eqs.8 and 9) [52,53].The SO52−reacted with H2O to produce O2•−(Eq.21) [32].The O2•−and SO5•−further reacted with•OH/H+and H2O to generate1O2(Eqs.22–24)[26,56].

OII mineralization was evaluated to provide insight into the degradation of OII in the system.The total organic carbon (TOC)removal rate was 11.2% and 36.6% at 10 min and 60 min (Fig.S8 in Supporting information), respectively.The OII was partially removed from the system, of which a small fraction was completely mineralized.Therefore, the degradation and intermediate OII products may remain as residual substances.Gas chromatography mass spectrometry (GCMS) was used to analyze the reaction intermediates and elucidate the degradation pathway of OII in the GBO + PMS system.Table S3 (Supporting information) shows that twelve degradation intermediates were detected.The potential OII degradation pathways are proposed in Fig.S9 (Supporting information) according to the reaction mechanism and identified intermediates.The degradation process was initiated by N=N bond cleavage due to the oxidative attack of•OH, SO4•−, and O2•−radicals, as well as1O2, leading to the formation of 1-amino-2-naphthol and sodium sulfanilamide [28,57–59].The 1-amino-2-naphthol was oxidized to form 1,2-dihydroxynaphthalene and 1-methylnaphthalene[59], while sodium sulfanilamide oxidation resulted in toluene,pxylene, andp-benzoquinone.All the intermediates were further oxidized and mineralized to CO2, H2O, NO3−, and SO42−[7].
In summary, this work highlights the outstanding ability of GBO to catalyze PMS decomposition, removing 92.2% of OII within 60 min.An exceedingly lowEalevel is achieved for OII degradation in the GBO + PMS system.The established GBO + PMS system shows significant potential for degrading OII in a broad pH range.1O2is identified as the primary reactive species for OII degradation, followed by•OH, SO4•−, and O2•−.Moreover, the results indicate that the Mn and Fe species play a crucial role in catalytic activity, while GBO also exhibits excellent stability and reusability.This work provides a novel application approach for GBO as an efficient PMS activator and will be useful during the future design and fabrication of transition metal PMS catalysts.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors acknowledge the funding support from Talent Introduction Program of Xihua University (No.Z202117).The authors would like to thank Shiyanjia Lab (www.shiyanjia.com) for the ESR analysis.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.01.029.
杂志排行

Chinese Chemical Letters的其它文章
- Zeolite-based Fenton-like catalysis for pollutant removal and reclamation from wastewater
- 1,n-Thiosulfonylation using thiosulfonates as dual functional reagents
- Degradation of florfenicol in a flow-through electro-Fenton system enhanced by wood-derived block carbon (WBC) cathode
- Simultaneous determination of indole metabolites of tryptophan in rat feces by chemical labeling assisted liquid chromatography-tandem mass spectrometry
- Self-powered anti-interference photoelectrochemical immunosensor based on Au/ZIS/CIS heterojunction photocathode with zwitterionic peptide anchoring
- The role of Cs dopants for improved activation of molecular oxygen and degradation of tetracycline over carbon nitride