中国罕见病药物临床试验10年现状分析:基于《第一批罕见病目录》
2022-12-05韩晓红
陈 晨,韩晓红
中国医学科学院北京协和医院 1临床药理研究中心 国家药监局药物临床研究与评价重点实验室创新药物临床PK/PD北京市重点实验室 2疑难重症及罕见病国家重点实验室,北京 100730
罕见病目前尚无统一的国际定义[1],其是一类发病率极低、多于儿童期起病、常危及生命的疾病。虽然罕见病单一病种患病人数少,但由于种类多、人口基数大,我国总体患病人数仍然较多。2018年5月,我国国家卫生健康委员会、科学技术部、工业和信息化部、国家药品监督管理局和国家中医药管理局联合发布了《第一批罕见病目录》,涵盖了121种罕见病[2]。
治疗罕见病的药物被认定为孤儿药。截至2022年8月,用于治疗121种罕见病的药物在美国、欧盟、日本已上市的药物涉及86种罕见病,而这86种罕见病中,仍有9种未在我国上市,且我国罕见病治疗药物主要依赖于进口[3]。为解决我国罕见病患者的用药问题,孤儿药的本土化生产日益受到重视,其中瓶颈问题之一是孤儿药研发过程中的药物临床试验[4]。因此,分析我国罕见病药物临床试验发展现状和研究态势具有重要意义。
本研究基于国家药品监督管理局药物临床试验登记与信息公示平台,汇总和分析了2012—2021年《第一批罕见病目录》涵盖的121种罕见病药物临床试验情况,旨在通过具体的数据分析,探讨罕见病药物临床试验现状与特点,为完善我国罕见病药物临床试验研究相关决策提供数据支持。
1 资料与方法
1.1 数据来源
本研究为回顾性分析。数据来自国家药品监督管理局药物临床试验登记与信息公示平台(http://www.chinadrugtrials.org.cn)登记数据库。以《第一批罕见病目录》涵盖的121种罕见病为主检索词,同时以《罕见病诊疗指南》中121种罕见病别名或细分疾病名称为补充检索词检索该数据库2012年11月1日(开放注册)至2021年11月28日登记的罕见病药物临床试验相关信息。
1.2 数据提取
从最终纳入研究的罕见病相关药物临床试验登记信息中提取数据信息,主要包括:(1)基本信息:题目、申办者、主要研究者、公示日期等;(2)试验科学信息:目的、试验药物、适应证、试验设计(包括试验分期、设计类型、试验范围)等注册试验的相关特征。
1.3 试验分期及归类
根据药物临床试验登记与信息公示平台中各临床试验详细信息中登记的试验分期,将Ⅰ期、Ⅰb/Ⅱ期、1/2/3期、Ⅰb/Ⅱa期和Ⅰ/Ⅱa期均划分为Ⅰ期临床试验;Ⅱ期和Ⅱ/Ⅲ期划分为Ⅱ期临床试验;药代动力学试验、探索性临床试验等划分为其他。
1.4 统计学处理
采用Excel 2020软件录入、整理数据并进行描述性分析,计数资料采用频数(百分数)表示。临床试验年均增长率(%)={2021年的临床试验数目/有临床试验计数的最早年份的临床试验数目)^[1/(2021-有计数最早的年份)]-1}×100%。
2 结果
2.1 近10年我国罕见病药物临床试验发展总体趋势
2012年11月1日至2021年11月28日,国家药品监督管理局药物临床试验登记与信息公示平台共登记15 134项注册临床试验,筛选出235项《第一批罕见病目录》涉及的罕见病药物临床试验。其中Ⅰ期41项,Ⅱ期22项,Ⅲ期74项,Ⅳ期15项,生物等效性试验77项,其他6项。从发展趋势看,我国罕见病药物临床试验数目总体呈逐年增长态势,年均增长率约为54%。此外,我国开展罕见病生物等效性试验较多,占比约为33%(表1,图1)。

图1 2012—2021年《第一批罕见病目录》中罕见病药物临床试验分布及趋势

表1 2012—2021年中国《第一批罕见病目录》中罕见病临床试验分布[n(%)]
2.2 Ⅰ/Ⅱ期临床试验涉及的罕见病领域
Ⅰ期和Ⅱ期罕见病药物临床试验共涉及16种罕见病。其中Ⅰ期临床试验涉及10种罕见病,主要集中于血友病、特发性肺纤维化、多发性硬化症和视神经脊髓炎谱系疾病;涉及33种药物,其中国产创新药20种、国产非创新药7种、跨国企业药物6种。Ⅱ期临床试验涉及11种罕见病,主要集中于特发性肺纤维化、视神经脊髓炎谱系疾病;涉及20种药物,其中国产创新药9种、国产非创新药4种、跨国企业药物7种(表2)。
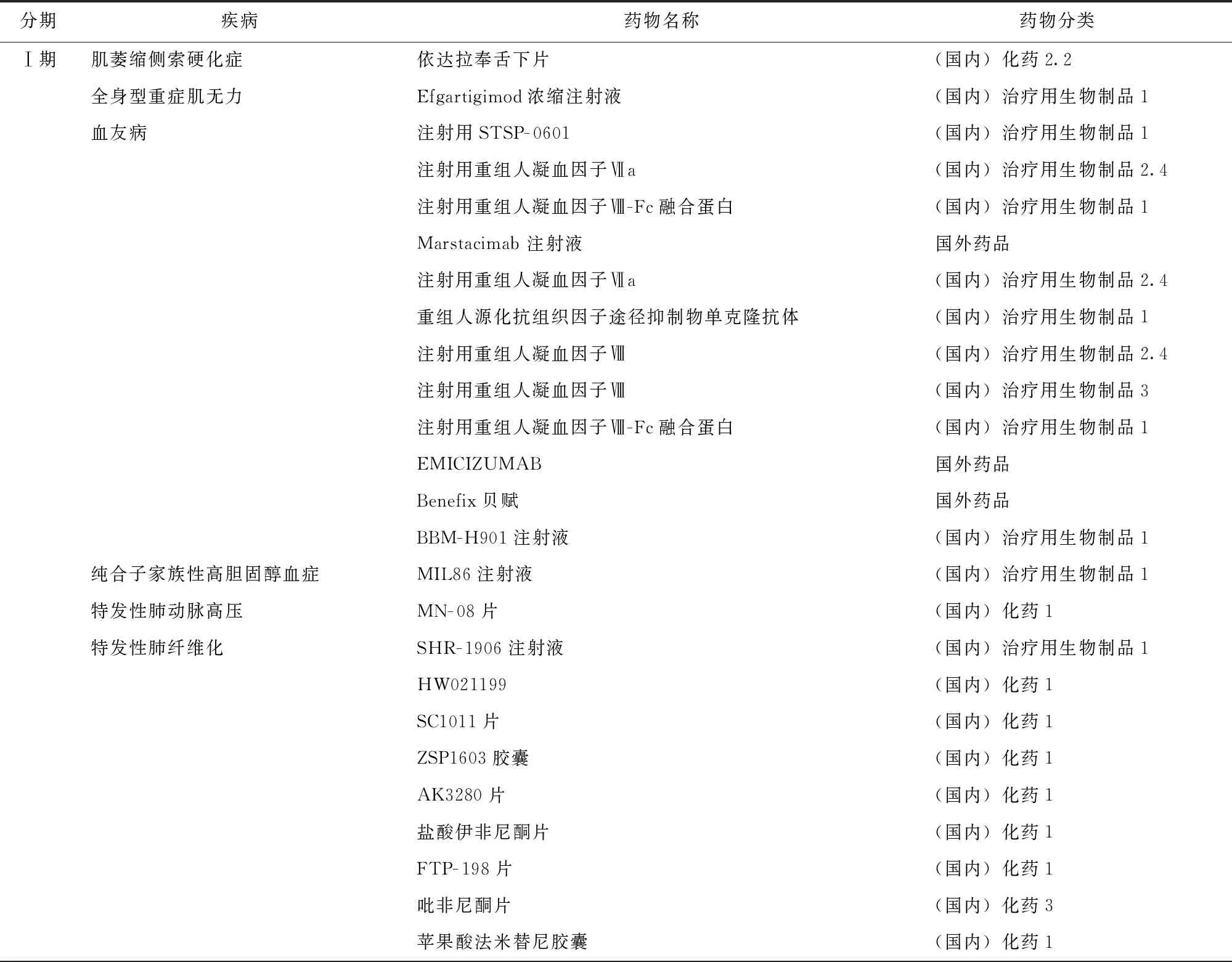
表2 2012—2021年《第一批罕见病目录》中罕见病Ⅰ/Ⅱ期药物临床试验涉及的疾病和药物
2.3 各期临床试验主要研究者地域分布特征
目录中包含的罕见病药物临床试验主要研究者地域分布广泛,共涵盖26个省/市(自治区)。其中,北京市研究者牵头的临床试验最多,Ⅰ期14项、Ⅱ期17项、Ⅲ期23项;其次是天津市,Ⅰ期12项、Ⅲ期18项;上海市排名第三,Ⅰ期9项、Ⅱ期6项、Ⅲ期4项(图2)。
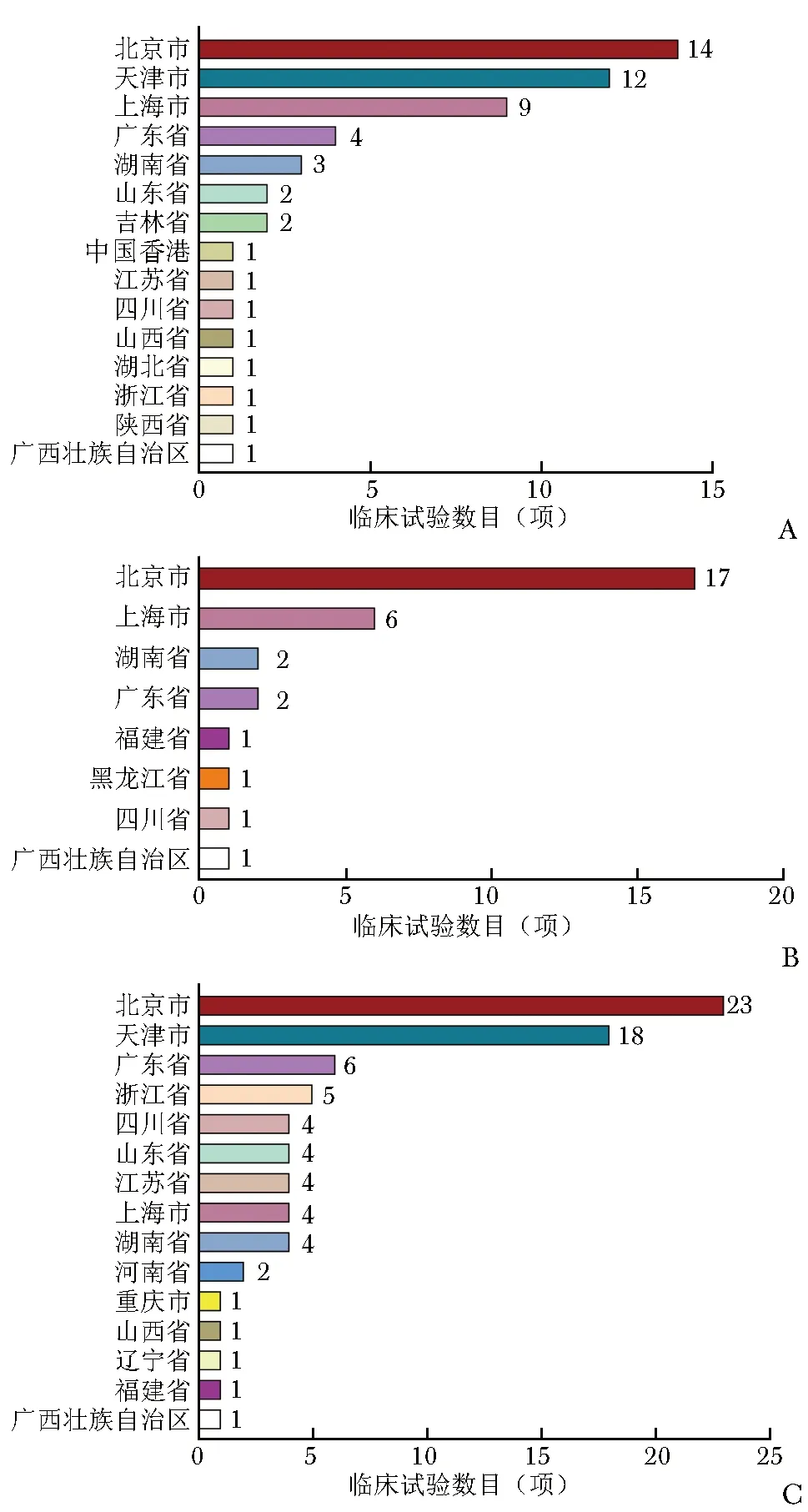
图2 2012—2021年《第一批罕见病目录》中罕见病药物临床试验主要研究者地域分布
2.4 各期临床试验设计类型
41项Ⅰ期临床试验中,国内单中心临床试验25项(61%),国内多中心临床试验16项(39%)。Ⅱ期临床试验中,国内单中心临床试验1项,国内多中心临床试验有12项(55%),国际多中心临床试验9项(41%)。Ⅲ期临床试验以国内多中心为主,共45项(61%),国际多中心临床试验共28项(38%)。平行对照研究设计Ⅰ期19项(46%),Ⅱ期13项(65%),Ⅲ期25项(34%);单臂试验Ⅰ期15项(34%),Ⅱ期8项(36%),Ⅲ期47(64%);交叉研究Ⅰ期7项(20%),Ⅱ期1项(5%),Ⅲ期2项(3%),见表3。

表3 2012—2021年《第一批罕见病目录》中罕见病药物临床试验设计情况[n(%)]
2.5 各期临床试验受试者规模
从受试者规模来看,Ⅰ期临床试验受试者人数主要集中于11~50例(83%,34/41),仅1项受试者人数<10例;受试者规模随分期增加逐渐增大,Ⅱ期13项(59%,13/22)大于50例,其中51~100例5项,>100例8项;Ⅲ期 51~100例21项(28%,21/74),101~500例18项(24%,18/74),>500例8项(11%,8/74),详见表4。

表4 2012—2021年《第一批罕见病目录》中罕见病药物临床试验受试者规模[n(%)]
3 讨论
本研究对国家药品监督管理局药物临床试验登记与信息公示平台开放注册以来至2021年11月28日,中国《第一批罕见病目录》中涵盖的121种罕见病药物临床注册试验进行了统计分析,结果显示我国罕见病药物临床试验开展总数较少,且新药研发仅涉及10余种罕见病。但近10年来罕见病药物研发呈快速增长趋势,临床试验注册年均增长率高达54%。
我国罕见病药物临床试验的快速增长得益于我国为促进罕见病药物研发出台的一系列政策[5]。其一是优先及加速审评审批,如2015年8月,国务院印发《关于改革药品医疗器械审评审批制度的意见》,明确加快审评审批罕见病等疾病的创新药;2015年11月,国家药监局发布《关于药品注册审评审批若干政策的公告》,指出罕见病等疾病的创新药注册申请实行单独排队,加快审评审批。此外,针对孤儿药研发建立专门的保护制度,2018年4月26日,国家药品监督管理局办公室公开征求《药品试验数据保护实施办法(暂行)》意见中提出,罕见病用药自该适应证首次在中国获批之日起给予6年数据保护期。但目前,我国与国外罕见病药物研发仍存在一定差距。一项既往研究结果显示,截至2019年10月底,基于ClinicalTrials.gov平台检索的481项针对罕见病的药物临床试验中,美国注册的罕见病临床试验最多,共233项,占48%[6]。中、美两国罕见病药物临床试验的数目悬殊主要是由于我国生物医药研究起步较晚,尚处于成长期,仍存在基础研究水平低、自主创新能力相对较弱等问题,因此对孤儿药的研发也相应落后[7]。
本研究发现,《第一批罕见病目录》中的121种罕见病相关药物的早期临床试验涉及的疾病类型仅有16种,主要集中于血友病、特发性肺纤维化、多发性硬化症和视神经脊髓炎谱系疾病。这一现象首先与罕见病发病率相关,发病率高、患病人数多的罕见病,易于招募受试者和开展相关临床试验。一项针对我国三级甲等医院住院病例(1500万例次)中涉及罕见病情况的调查结果显示,居于前三的罕见病为特发性肺动脉高压、特发性肺纤维化、多发性硬化症[8],反映出我国此类罕见病发病率相对较高。而由于121种罕见病中有58种累及对象是儿童,进一步限制了潜在临床试验参与者的可获得性[9]。其次,药物研发进程还与对疾病发病机制和致病机理的认识程度有关,靶点机制研究越深入,针对不同靶点的新药研究进程也越快。如血友病的发病机制和分型已非常明确,按照患者所缺乏的凝血因子类型可快速进行药物研发,截至2021年11月,美国食品药品监督管理局(Food and Drug Administration,FDA)已批准18种针对血友病的孤儿药上市。美国临床试验数据库平台数据显示,神经鞘脂病、范科尼综合征、卟啉症等相关罕见病临床试验相对较多[6],这与本研究结果存在明显差异,而罕见病药物临床试验涉及领域的差异从侧面反映了我国罕见病疾病谱的不同,因此需制订适合我国国情的药物研发策略。
本研究还发现,罕见病药物早期临床试验中,平行对照试验设计最多,其次是单臂开放试验,说明我国罕见病药物临床试验依然以传统临床试验设计为主,同时也提示平行对照试验在发病人数相对较多的罕见病早期临床试验中具有可行性。尽管随机对照试验一般被认为是评价药物安全性和有效性的金标准,并为普通疾病药物临床研究普遍采用,但本研究中64%的罕见病Ⅲ期药物临床试验采用的是单臂试验设计,进一步说明罕见病药物临床试验设计不同于普通疾病。对于某些缺乏有效治疗措施的罕见病,考虑其疾病特殊性和阳性对照药物的合理性问题,可采用真实世界证据作为其外部对照。此外,罕见病药物临床试验需根据不同阶段的特点采用灵活的替代临床试验设计(包括适应性无缝试验、交叉试验、随机安慰剂设计、随机撤药设计、多臂试验、平台试验、篮子试验等[10]),以通过有限的患者数据,获得满足获益与风险评估的科学证据,提高药物研发效率[11]。例如,2020年8月7日,美国FDA基于关键性临床FIREFISH研究(针对2~7个月患儿)和SUNFISH研究(针对2~25岁患者)的数据批准利司扑兰(risdiplam)上市,用于治疗成人和2个月以上儿童脊髓性肌萎缩症(spinal muscular atrophy, SMA)。其中FIREFISH研究是一项开放性、两部分无缝衔接、多中心Ⅱ/Ⅲ 期临床研究。第一部分为开放、非随机剂量递增研究,共入组21人;第二部分为疗效验证研究,共入组41人,在第一部分选择的剂量下口服治疗24个月后,进入为期3年的开放延长期[12- 13],这一无缝试验加速了剂量选择验证过程,为药物上市节省了时间。又如,一种治疗儿童和成人Ⅶ型黏多糖贮积症(mucopolysaccharidosis,MPS Ⅶ)的重组人β葡糖醛酸糖苷酶药物Mepsevii,其Ⅰ/Ⅱ期临床试验在初始治疗期后进行强制剂量滴定,以评估最佳剂量, Ⅲ期临床试验采用随机双盲、自身安慰剂对照以及自身交叉设计,同时采用多域反应指数(multi-domain responder index, MDRI)为主要终点指标,Ⅰ~Ⅲ期临床试验共入组23名受试者,基于此有限患者的临床试验结果,美国FDA批准了该药上市[14- 15]。未来,在我国罕见病药物临床试验中,应通过采用灵活的试验设计,从有限的患者数据,获得满足获益与风险评估的科学证据,提高罕见病药物临床试验效率。
基于国家权威数据库,本研究从多角度分析了我国罕见病药物临床试验的现状和特点,但仍存在一定局限性:(1)数据来源较单一,仅包含了国家药品监督管理局药物临床试验登记与信息公示平台的数据,其他平台(如中国临床试验注册中心)注册的由研究者发起的罕见病药物临床试验研究并未纳入分析,结果可能存在一定偏倚。(2)本研究纳入分析的是药物临床试验公示日期,与既往研究中基于的首次伦理审查日期有所延后,与试验实际开展时间亦稍有差异。
综上,我国罕见病药物研发尚处于发展初期阶段,在国家政策的支持和鼓励下,本土制药企业发展迅速,自主创新能力正在逐渐加强,未来我国罕见病药物临床试验必将实现更大的突破,惠及更多患者。
作者贡献:陈晨负责论文初稿撰写、数据整理分析及修订;韩晓红负责研究设计、论文审阅和修改。
利益冲突:所有作者均声明不存在利益冲突
